
Abstract
Intestinal epithelial cells (IECs) are the primary target of enteric viruses. Their infection by viruses leads to the upregulation of both type I and type III interferons (IFNs). These IFNs then act in an autocrine and paracrine manner to protect IECs from viral propagation. To date, whether both IFNs use similar signaling pathways and whether these 2 cytokines can act synergistically to protect against viral infection remain unclear. Using human IECs depleted of either the type I or type III IFN receptor, we found that both signal transduction pathways are interconnected and influence each other at the level of interferon-stimulated gene (ISG) expression and efficiency of antiviral protection. Precisely, in human IECs, the presence of a functional type III IFN receptor negatively regulates type I IFN signaling and activity, whereas the presence of type I IFN receptor positively reinforces type III IFN signaling and function. We propose that this complex crosstalk allows for a preferential type III IFN-mediated protection of human intestinal cells.
Introduction
Interferons (IFNs) are cytokines with pleiotropic functions ranging from effects on cellular physiology such as the regulation of cell survival, differentiation and proliferation, protein translation and metabolism to regulation of diseases such as autoimmune or autoinflammatory conditions, cancer, and, most importantly, the regulation of viral and nonviral pathogenic infections (Gibbert and others 2013; Schneider and others 2014).
The IFN family of cytokines is divided into 3 types: I, II, and III based on common features in gene sequence, protein structure, expression pattern, and receptor engagement. While the type II IFN family, consisting of a sole member (IFN-γ), acts as an immunomodulatory cytokine, type I IFNs, consisting of multiple subtypes: IFN-α(1–13), IFN-β, IFN-ɛ, IFN-κ, and IFN-ω, and type III IFNs, consisting of 4 members: IFN-λ1 [interleukin-29 (IL-29)], IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and IFN-λ4, act as the main antiviral mediators in mammals (Isaacs and others 1957; Dumoutier and others 2003; Kotenko and others 2003; Sheppard and others 2003; Prokunina-Olsson and others 2013).
Type I and type III IFNs signal through different heterodimeric receptor complexes, which importantly present differential localization reflecting a compartmentalized function. The type I IFN receptor complex consists of the IFNαR1 and IFNαR2 chains, and the type III IFN receptor complex is composed of the IFNλR1 and IL-10RB chain (Gibbert and others 2013). The IFNαR1, IFNαR2, and IL-10RB subunits are broadly distributed in various cell types, whereas the expression of the IFNλR1 chain is predominately restricted to epithelial cells, neutrophils, and keratinocytes (Witte and others 2009; Blazek and others 2015; Broggi and others 2017). The expression of the type III IFN receptor in epithelial cells (Kotenko and others 1997, 2003; Sheppard and others 2003; Sommereyns and others 2008) provides this cytokine an important epitheliotropic function. In particular, while type III IFNs mainly protect the epithelial cells of the respiratory, gastrointestinal, and urogenital tract, type I IFNs contribute to control systemic responses responsible for inhibiting viral dissemination beyond the mucosal barrier (Pott and others 2011; Mahlakõiv and others 2015; Nice and others 2015; Lin and others 2016; Klinkhammer and others 2018).
Upon viral infection, type I and type III IFN production is mediated by pathogen-recognition receptors (PRRs), which sense a broad range of pathogen-associated molecular patterns (PAMPs) including viral ligands, such as nucleic acids (Schneider and others 2014; Hoffmann, and others 2015). Upon production, type I and type III IFNs are secreted in the extracellular environment where they act in an autocrine and paracrine manner to induce their antiviral activity through the expression of hundreds of interferon-stimulated genes (ISGs) responsible for the restriction of viral infection in both infected and noninfected cells (Kotenko and Durbin, 2017).
Binding of IFNs to their receptors induces the activation of members of the receptor-associated Janus kinase (JAK) family, which phosphorylate tyrosine residues on the receptor molecules (Zhou and others 2007; Lavoie and others 2011; de Weerd and Nguyen 2012). This phosphorylation mediates the recruitment of signal transducer and activator of transcription (STAT) proteins, which are then phosphorylated by JAKs. STAT activation results in dimerization of STATs and the formation of the IFN-stimulated gene factor 3 (ISGF3) complex, consisting of STAT1, STAT2, and IRF9. Translocation of the ISGF3 or dimers of STATs to the nucleus regulates the expression of ISGs (de Weerd and Nguyen 2012).
Taking into account that type I and type III IFNs utilize the same signaling cascade and induce almost the same pool of ISGs, it was originally proposed that both IFNs have redundant functions. However, a number of signaling and functional differences have been recently discovered and are associated with diverse antiviral and immunoregulatory activity of type I versus type III IFNs (Mordstein and others 2008, 2010; Pott and others 2011; Baldridge and others 2015; Mahlakõiv and others 2015; Nice and others 2015; Lin and others 2016; Klinkhammer and others 2018). While the MAPKinase pathway is activated downstream of both IFN receptors, it has been shown to contribute specifically in the antiviral activity of type III IFNs in intestinal epithelial cells (IECs) (Pervolaraki and others 2017). Furthermore, important quantitative differences have been demonstrated in the magnitude and kinetics of induction of ISGs between type I and type III IFNs in different epithelial cell lines and ex vivo models of intestinal organoid cultures, where type I IFNs trigger a stronger and faster induction of ISGs compared with a more delayed and reduced ISG response stimulated by type III IFNs (Marcello and others 2006; Bolen and others 2014; Jilg and others 2014; Bhushal and others 2017; Pervolaraki and others 2017, 2018). Additionally, it was also recently determined that type III IFNs display an intestinal-specific induction of a distinct subset of ISGs further supporting the model of distinct functions of the 2 IFNs at epithelial surfaces (Selvakumar and others 2017).
Although it is becoming more documented that there are differences in the signal transduction pathways regulating the production of ISGs and the function of type I versus type III IFNs, it remains unclear whether there is a crosstalk occurring between the signaling pathways downstream of the type I and type III IFN receptors. To directly address whether the presence of a functional type I IFN-mediated signaling cascade can influence type III IFN signaling/function and vice versa, we exploited IECs where either the type I or type III IFN signaling pathways have been ablated. Using this method, we determined that the lack of either type I or type III IFN receptors influenced the IFN-mediated production of ISGs and antiviral function of the reciprocal IFN.
Materials and Methods
Antibodies/reagents
Commercially available primary antibodies were mouse IgG1 kappa (No. 14-4714-82, clone P3.6.2.8.1; Invitrogen) monoclonal antibodies recognizing beta-actin (No. A5441; Sigma), phospho STAT1 and STAT1 (No. 612233 and No. 610115, respectively; BD Transductions), anti-mouse antibodies (No. NA934V; GE Healthcare) coupled with horseradish peroxidase were used as secondary antibodies for Western blot at a 1:5,000 dilution. In the neutralization assays, the mouse monoclonal antibodies against human IFN alpha receptor 2 (IFNAR2; clone: MMHAR2, pbl No. 21385-1) and human IFN lambda receptor 1 (IFNLR1; clone: MMHLR-1, pbl No. 21885-1) were used. Human recombinant IFN-beta1a (IFN-β) was obtained from Biomol (No. 86421). Recombinant human IFN-λ1 (IL-29; No. 300-02L) and IFN-λ2 (IL-28A; No. 300-2K) were purchased from PeproTech and IFN-λ3 (IL-28B) from Cell Signaling (No. 8796). The IFN concentrations used to treat the cells are stated in the figure legends.
Cell and viruses
Wild-type (WT) and IFN receptor knockout T84 human colon carcinoma cells (ATCC CCL-248) were maintained in a 50:50 mixture of Dulbecco's modified Eagle's medium and F12 (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Gibco). WT and IFN receptor knockout cells were seeded at similar densities as they show no difference in their growth or polarization rates. Vesicular stomatitis virus expressing a luciferase reporter (VSV-luc) was prepared and used as previously described (Pervolaraki and others 2018).
RNA isolation, complementary DNA, and quantitative polymerase chain reaction
RNA was harvested from cells using NucleoSpin RNA extraction kit (Macherey-Nagel) as per the manufacturer's instructions. Complementary DNA (cDNA) was made using iSCRIPT reverse transcriptase (Bio-Rad) from 250 ng of total RNA as per the manufacturer's instructions. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using SsoAdvanced SYBR green (Bio-Rad) as per the manufacturer's instructions; TBP and HPRT1 were used as normalizing genes.
Western blot
At time of harvest, media was removed, cells were rinsed 1 time with 1 × phosphate-buffered saline (PBS) and lysed with 1 × RIPA buffer [150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris, pH 8.0 with phosphatase and protease inhibitors (Sigma-Aldrich)] for 20 min at 4°C. Lysates were collected and equal protein amounts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and blotted onto a nitrocellulose membrane by wet-blotting (Bio-Rad). Membranes were blocked with 5% milk or 5% bovine serum albumin (BSA), when the phospho STAT1 antibody is used, in TBS containing 0.1% Tween 20 (TBS-T) for 1 h at room temperature (RT). Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. Membranes were washed 4 × in TBS-T for 15 min at RT. Secondary antibodies were diluted in blocking buffer and incubated at RT for 1 h with rocking. Membranes were washed 4 × in TBS-T for 15 min at RT. HRP detection reagent (GE Healthcare) was mixed 1:1 and incubated at RT for 5 min. Membranes were exposed to film and developed.
VSV-luc assay
T84 cells were seeded in a white F-bottom 96-well plate. Cells were treated as indicated in the main text or figure legends with type I or type III IFNs in the presence or absence of neutralizing antibodies. VSV-luc was added to the wells and the infection was allowed to proceed for 8 h. At the end of the infection, media was removed, cells were washed 1 × with PBS and lysed with Cell Lysis Buffer (Promega) at RT for 20 min. A 1:1 dilution of Steady Glo (Promega) and Lysis Buffer were added to the cells and incubated at RT for 15 min. Luminescence was read using an Omega Luminometer.
Luminex assay
At time of harvest, media was removed, cells were rinsed 1 time with 1 × PBS and lysed with 1 × MILLIPLEX MAP Lysis buffer (catalog No. 43-040; EMD Millipore) supplemented with protease inhibitors (Sigma–Aldrich) for 20 min at 4°C. Lysates were collected, filtered (catalog No. UFC30DV00; EMD Millipore), and equal protein amounts were used to detect the phospho STAT1 presence using the phosho STAT1 (Tyr701) Magnetic Bead Mapmate kit (catalog No. 46-655MAG; EMD Millipore) according to the manufacturer's instructions.
Results
Type I and type III IFN receptor-specific knockouts alter ISG expression
Given the similarities in the signaling pathways downstream the type I and type III IFN receptor complexes, we wondered whether both receptors could influence each other's signal transduction and antiviral function through an inter-receptor molecular crosstalk. To test for a functional crosstalk between type I and type III IFN signaling pathways, we examined the transcriptional activity of IFNs in the human colon carcinoma-derived cell line T84 deficient for either the IFN alpha receptor 1 (IFNAR1−/−) or the IFNLR1−/− (Pervolaraki and others 2017). IFNAR1−/− T84 cells displayed an increased base line expression of ISGs (IFIT1 and Viperin) compared with WT cells (Fig. 1A, B). On the contrary, IFNLR1−/− showed a strongly reduced base line expression of ISGs (Fig. 1A, B). WT and receptor knockout cells were treated with type I or type III IFNs, and the expression levels of 2 representative ISGs (IFIT1 and Viperin) were assayed at different times post-IFN treatment. IFNAR−/− cells show reduced levels of both IFIT1 and Viperin expression particularly at 24 h post-IFN treatment (Fig. 1C, D). Contrarily, IFNLR1−/− cells displayed an increase in the levels of ISG induction upon type I IFN treatment at their peak of expression compared with WT cells (Fig. 1E, F). Notably, there is no difference in the kinetic pattern of ISG induction in IFNAR1−/− or IFNLR1−/− cells compared with WT cells upon treatment with type III or type I IFN, respectively. In all cell lines, type III IFN treatment induces a delayed expression of ISGs with a lower magnitude of ISG expression compared with type I IFN, as previously reported (Marcello and others 2006; Bolen and others 2014; Jilg and others 2014; Bhushal and others 2017; Pervolaraki and others 2017, 2018). Importantly, when comparing ISG induction upon IFN treatment with steady-state levels, results show that IFNAR1−/− cells were less responsive to type III IFN treatment compared with WT cells expressing both IFN receptors (Supplementary Fig. S1A, B), whereas IFNLR1−/− cells were significantly more responsive to type I IFN compared with WT cells (Supplementary Fig. S1C, D).
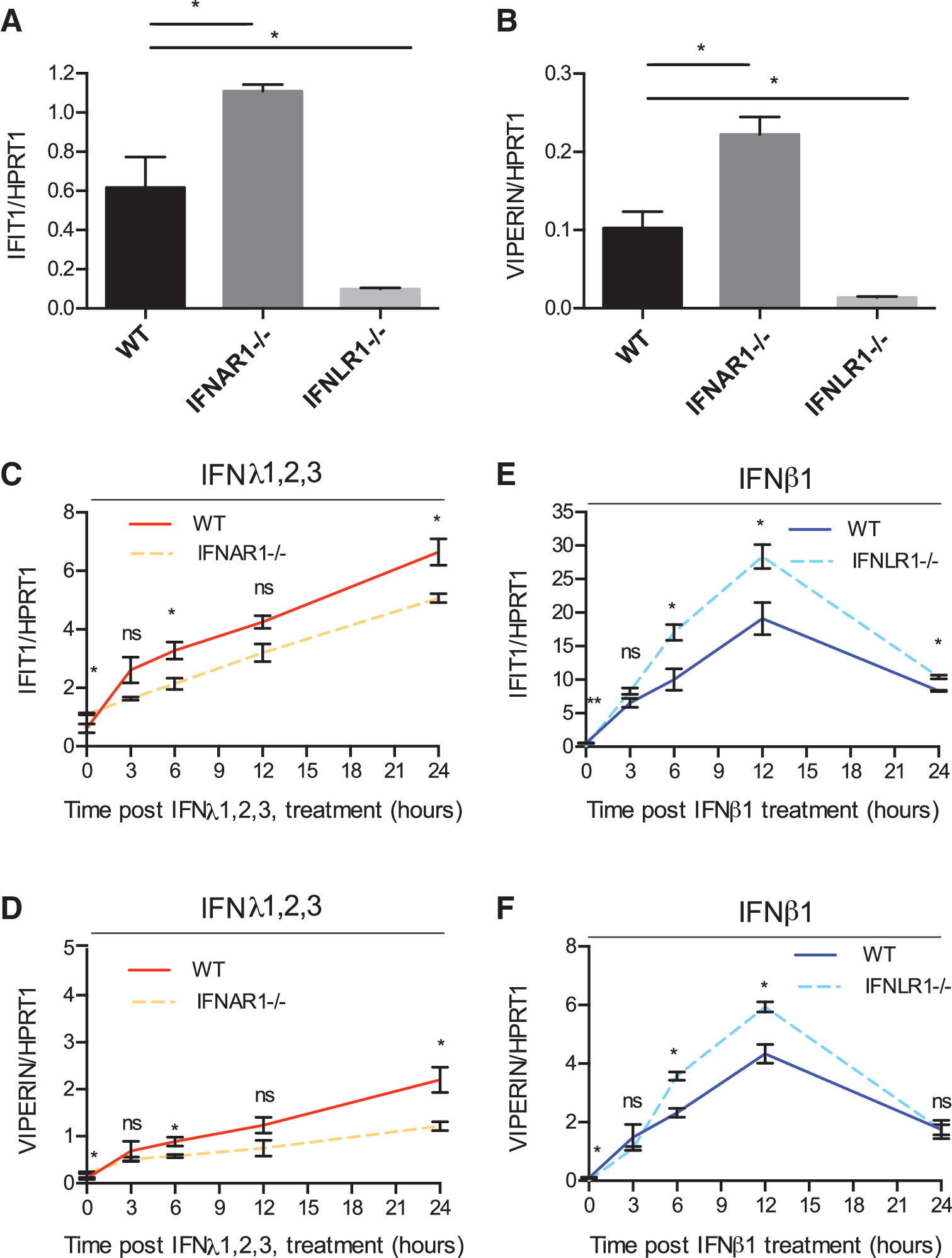
IFN receptor knockouts show differential levels of ISG expression.
To directly control that this difference in ISG expression was the result of the knockout of the individual IFN receptors, we performed rescue experiments where CRISPR/Cas9 guide RNA-resistant IFNAR1 or IFNLR1 constructs were transduced into the corresponding IFN receptor knockout cells. As we previously reported, overexpression of the IFN receptors in the corresponding knockout lines resulted in a rescue of both the ISG expression and antiviral function upon IFN treatment (data not shown and Pervolaraki and others 2018). Importantly, rescue of IFNAR1 in the IFNAR1−/− cells resulted in an increase of ISG production upon type III IFN treatment (Fig. 2A, B). Similarly, rescue of IFNLR1 in IFNLR1−/− cells resulted in a decrease of ISG expression upon type I IFN treatment (Fig. 2C, D). Taken together, these results strongly suggest that there is a functional crosstalk between the signaling pathways downstream of both type I and type III IFNs, where the type I IFN receptor acts in a positive manner on type III signaling while the type III IFN receptor acts as a negative regulator of type I IFN signaling.

Transcomplementation of IFN receptor in IFN receptor knockout cells rescue ISG expression.
IFN receptor-specific neutralizing antibodies reveal a functional crosstalk between type I and type III IFNs
To address whether the differential induction of ISGs seen in the IFN receptor knockout lines could be recapitulated through acute inhibition of IFN signaling, we made use of neutralizing antibodies against each IFN receptor. First, we began by controlling the functionality of each neutralizing antibody, we titrated the IFNAR2 neutralizing antibody with increasing concentrations of type I IFN and the IFNLR1 neutralizing antibody with increasing concentrations of type III IFN to determine the amount of antibody/IFN combination required to reach a level of 50% inhibition of ISG levels (data not shown). Then, T84 WT cells were treated with either the IFNAR2 or IFNLR1 antibody before stimulation with type I or type III IFN, respectively (Fig. 3A). Pretreatment of cells with the neutralizing antibody leads to a significant decrease in ISG production upon type I IFN treatment (Fig. 3B) or type III IFN treatment (Fig. 3C). To determine whether this decreased ISG production correlated with a reduced antiviral function, WT cells were mock treated or treated with the IFNAR2 or IFNLR1 neutralizing antibody before pretreatment of cells with type I or type III IFN, respectively, before infection by VSV-luc (Fig. 3D). As expected, the inhibition of ISG production (Fig. 3B, C) correlated with an increased permissiveness of the cells during viral infection (Fig. 3E, F). In addition, treatment of T84 cells with an isotype control IgG antibody did not alter ISG expression or antiviral protection following type I or type III IFN treatment (Fig. 3B, C, E, F). Interestingly, inhibition of the type III IFN receptor leads to an increase in type I IFN-mediated ISG induction and a moderate increase in type I IFN-mediated antiviral activity (Fig. 3B, E). Conversely, inhibition of type I IFN signaling resulted in a significant reduction in the level of ISG expression upon type III IFN treatment (Fig. 3C). Importantly, this was associated with a partial loss of protection mediated by type III IFN as can been seen by an increase in VSV-luc infection (Fig. 3F). This shows that the type I IFN receptor positively regulates type III IFN-mediated induction of ISGs and antiviral function, whereas the type III IFN receptor negatively regulates type I IFN-mediated ISG induction and antiviral properties.

Neutralizing antibody treatment of human IECs confirms the crosstalk between the type I and type III IFN receptors.
To validate the observed decrease of type III IFN-mediated signaling and increase of type I IFN-mediated signaling when using an anti-IFNAR2 and an anti-IFNLR1 neutralizing antibody, respectively, we controlled for the specificity of our neutralizing antibody. IFNLR1−/− cells were pretreated with the anti-IFNLR1 neutralizing antibody and then challenged with type I IFN. Similarly, IFNAR1−/− cells were pretreated with the anti-IFNAR2 neutralizing antibody and then challenged with type III IFN. Results show that both neutralizing antibodies were specific and did not affect ISG production or the antiviral function of the reciprocal IFN when treated in single receptor knockout cell lines (Supplementary Fig. S2A–D). Altogether, these results confirm our observation from the knockout cells showing the positive influence of the type I IFN receptor on type III IFN-mediated signaling and function as well as the negative crosstalk that the type III IFN receptor plays on type I IFN-mediated signaling.
Differential activity of type I and type III IFN signaling pathways is not due to different expression levels of the receptors
The observed increased activity of type I IFN signaling/activity in the absence or inhibition of type III IFN receptor could be explained by an increase of type I IFN receptor levels. Similarly, the decreased type III IFN-mediated signaling/activity in the absence or inhibition of type I IFN receptor could be due to a decrease of the IFNLR expression levels. To address these possibilities, we evaluated the levels of both IFN receptors in our WT, IFNLR1−/−, and IFNAR1−/− cells. Results show that the basal levels of IFNLR1 were unchanged in IFNAR1−/− cells (Fig. 4B), whereas the levels of IFNAR1 were slightly decreased in IFNLR1−/− cells compared with WT cells (Fig. 4A). Importantly, this reduction was only minimum and is unlikely to explain the observed increase of type I IFN-mediated signaling upon genetic ablation of IFNLR1 (Fig. 1E, F) or upon treatment of cells with an anti-IFNLR1 neutralizing antibody (Fig. 3B, E). Additionally, WT cells were treated with type I or type III IFN and the levels of IFNAR1 and IFNLR1 were controlled by qRT-PCR. Treatment of WT cells with either type I or type III IFN does not significantly change the expression levels of either IFNAR1 or IFNLR1 (Fig. 4A, B). Next, IFNLR1−/− cells were treated with type I IFN and the levels of IFNAR1 were measured by qRT-PCR. As with WT cells, there was no change in receptor level in the knockout cells in mock-treated conditions or upon addition of type I IFN (Fig. 4A). Similarly, IFNAR1−/− cells did not show a change in IFNLR1 levels in the presence or absence of type III IFN treatment (Fig. 4B). In addition, we did not observe a decrease in IFNLR1 expression upon treatment of cells with the IFNAR2 neutralizing antibody or an increase in IFNAR1 expression levels upon treatment of cells with the IFNLR1 neutralizing antibody (data not shown). Together, these results show that the differential ISG expression and antiviral properties observed upon IFN receptor knockout/inactivation are not due to changes in the reciprocal IFN receptor levels.
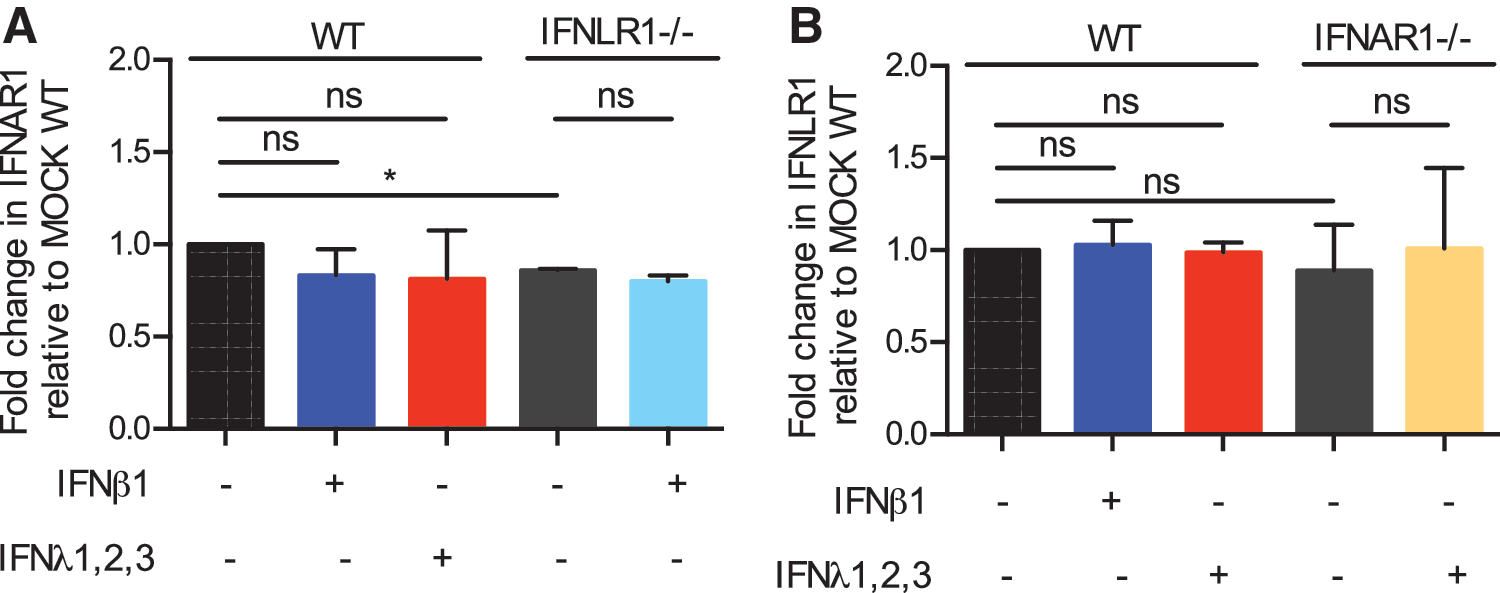
IFN receptor levels are not altered in reciprocal IFN receptor knockout cells.
Modulation of STAT1 activation drives the differential IFN-mediated signaling observed upon IFN receptor knockout
As JAK/STAT signaling molecules are the key players in the signaling pathway downstream type I and type III IFN receptors, we next wanted to test whether our observed crosstalk emanated from differential induction of STAT1. The phosphorylation kinetics of STAT1 was compared in IFNAR1−/− and WT cells upon treatment with type III IFN through Luminex assay. In line with the ISG induction (Fig. 1), weaker activation of STAT1, as seen by a decreased amount of STAT1 phosphorylation (p-STAT1), was observed in IFNAR1−/− cells compared with WT cells upon type III IFN treatment (Fig. 5A). Complementarily, Western blot analysis was used to further confirm the activation pattern of STAT1 and to investigate the total level of STAT1 in IFNAR1−/− cells. Similar to the Luminex results, the Western blot showed a decrease in p-STAT1 levels in IFNAR1−/− cells upon type III IFN treatment in comparison to WT cells (Fig. 5B–D). Quantification of the total levels of STAT1 revealed that there was no significant difference between WT and IFNAR1−/− cells in both mock- and type III IFN-treated cells (Fig. 5B, C and Supplementary Fig. S3A). In line with the IFNAR1−/− cells, WT cells treated with the neutralizing antibody against IFNAR2 showed a decrease in p-STAT1 levels upon type III IFN treatment (Supplementary Fig. S4A).

IFN receptor knockout influences the activation of STAT1.
Next, the phosphorylation kinetics of STAT1 was compared in IFNLR1−/− and WT cells upon treatment with type I IFN through Luminex assay. Elevated levels of p-STAT1 were detected in IFNLR1−/− cells treated with type I IFN (Fig. 5E), which correlates with the increased ISG levels detected after IFN treatment (Fig. 1). Western blot analysis further confirmed the increase in p-STAT1 levels upon type I IFN treatment of IFNLR1−/− cells compared with WT cells (Fig. 5F–H). Analysis of total STAT1 levels revealed no major differences between WT and IFNLR1−/− cells (Supplementary Fig. S3B). In line with these results, WT cells treated with the IFNLR1 neutralizing antibody displayed an increase in pSTAT1 levels upon type I IFN treatment (Supplementary Fig. S4B). Altogether, these results show a functional crosstalk between type I and type III IFN receptors where the type I IFN receptor positively influences type III IFN signaling by promoting p-STAT1 and conversely where type III receptor negatively influences type I IFN signaling by decreasing the amplitude of p-STAT1.
Discussion
In this work, we determined that a crosstalk occurs between the signal transduction pathways downstream the type I and type III IFN receptors allowing them to influence each other's IFN-mediated signaling. Using human IECs depleted of either the IFNαR1 or IFNλR1 receptor, we showed that ISG induction was altered when the cells were treated with the reciprocal IFN. Precisely, IFNAR1−/− cells lead to a decrease in ISG production after type III IFN treatment, whereas IFNLR1−/− cells lead to an increase in ISG production after type I IFN treatment. These observations were further confirmed when performing an acute inhibition of IFN-mediated signaling using neutralizing antibodies directed against either the type I or type III IFN receptors. This positive and negative crosstalk was not due to an upregulation or downregulation of the IFN receptor levels but due to a decreased and increased activation of STAT1. Our data show that the type I IFN receptor positively regulates type III IFN signaling, whereas the type III IFN receptor negatively influences type I IFN signaling.
Using type III IFN receptor knockout cell lines and neutralizing antibodies directed against IFNλR1, we could show that the presence of the type III IFN receptor negatively regulates both the production of ISGs and the antiviral state mediated by type I IFN (IFN-β) (Figs. 1 and 3). Previous work has shown that type III IFN was able to create a refractory state for IFN-α signaling in mice (François-Newton and others 2011; Makowska and others 2011). These studies show that pretreatment of cells with either IFN-β or IFN-λ induces the negative regulator USP18. This induction of USP18 was correlated with the decreased ability of IFN-α to activate STAT1 and promote ISG induction (François-Newton and others 2011; Makowska and others 2011). Importantly, in these studies, IFN-β signaling was not affected suggesting that even though both IFN-α and IFN-β use the same IFN receptor and share the same downstream JAK/STAT-dependent signaling cascade, they have different sensitivities to its negative regulation by USP18. Interestingly, the effect of USP18 seems to be more pronounced in the liver and is less apparent in intestinal tissues suggesting tissue-specific regulations of IFN signaling by USP18 (Makowska and others 2011). Here, using IECs, we can clearly demonstrate the presence of a negative regulatory mechanism downstream type III IFN receptor targeting type I IFN-mediated signaling. In the current work, we have not investigated the molecular origin of this negative regulatory loop, but since it impacts type I IFN-mediated signaling, it is tempting to speculate that USP18 might be involved. However, the fact that it efficiently impairs IFN-β signaling could argue for USP18-independent mechanism. Additional experiments exploiting USP18-deficient cells should be performed to challenge this hypothesis.
Opposite to the negative feedback that the type III IFN receptor generates on type I IFN-mediated signaling, our results highlight that the type I IFN receptor positively influences type III IFN-mediated production of ISGs and antiviral properties (Figs. 1 and 3). Type I IFNs have been previously described to display a positive feedback loop during viral infection. It was shown that Newcastle disease virus induced the IRF3-dependent production of the IFN-α4 subtype (Marie, and others 1998). This newly produced IFN-α4 was then secreted from the cells and through an IFNAR- and STAT1-dependent signaling cascade was able to drive the production of other IFN-α subtypes as well as IRF7, which together further propagated the IFN antiviral phenotype in a positive feedback loop. A similar positive feedback in type I IFN-mediated signaling has also been shown in splenocytes and vaginal tissue of IFNAR−/− mice, which displayed a decreased induction of both type I and type III IFN transcripts following herpesvirus or Sendai virus infection in comparison to WT mice (Ank and others 2008). This decreased IFN induction in IFNAR−/− splenocytes and vaginal cells is in line with the reduced ISG induction we see in our human IFNAR1−/− IECs (Fig. 1). These observations further support the idea that type I IFN has a positive feedback loop, which influences not only the production of more type I IFN but also type III IFN transcripts.
Interestingly, a recent study also demonstrated differences in the ability of mouse intestinal cells to respond to IFN-β and IFN-λ (Schwerk and others 2013). In this study, immortalized murine IECs and mouse organoids expressing an Mx2-RFP reporter were stimulated with either type I or type III IFN and the level of ISG induction was observed by fluorescence microscopy or florescence activated cell sorting (Bhushal and others 2017). It was shown that increasing levels of IFN-β continuously increased the amount of Mx2-RFP-positive cells within the population, whereas treatment of type III IFN, even at high doses, showed that only part of the cell population was capable of responding (Bhushal and others 2017). We and others have also observed this in human intestinal cell models where increasing concentrations of IFN-β lead to a continuous increase in ISGs while treatment of type III IFNs leads to a plateau of ISG induction at low concentrations (Saxena and others 2017; Pervolaraki and others 2018). These observations are compatible with the current study showing that type I IFN signaling leads to a positive feedback while type III IFNs do not.
Several groups have evaluated the effect of knocking out either the type I or type III IFN receptors in mice and have characterized how their absence affects enteric virus infection (Pott and others 2011; Mahlakõiv and others 2015; Nice and others 2015; Lin and others 2016). In these murine models, IFNλR1 expression is restricted to mostly IECs lining the gastrointestinal tract and IFNαR1 is expressed in the lamina propria of adult mice. Within the intestine, type III IFNs play a critical role in controlling infection of multiple enteric viruses (eg, rotavirus, reovirus, and mouse norovirus) within the epithelial cells, whereas type I IFNs are required to control their systemic dissemination (Pott and others 2011; Mahlakõiv and others 2015; Nice and others 2015; Lin and others 2016). Interestingly, most of these studies have not detected changes in the level of ISG induction upon enteric virus infections between WT and receptor knockout (Mahlakõiv and others 2015; Lin and others 2016). However, these studies have noticed that the basal levels of IFN transcripts are affected in knockout animals. These results are different from what we have found in our study. While we have also seen changes in the basal levels of IFN and ISG transcripts (data not shown and Fig. 1A, B), we see large changes in the induction of ISGs upon IFN treatment (Fig. 1 and Supplementary Fig. S1). A possible mechanism to explain the observed decrease of ISG expression upon type I IFN receptor knockout or neutralization is that inhibition of type I IFN-mediated signaling prevents the production of a basal level of type I IFNs, which are secreted in a constitutive manner by cells independent of infection. These ISGs could in turn promote a positive feedback on type III IFN-mediated signaling.
The discrepancy seen in IFN-mediated signaling upon IFNαR1 and IFNλR1 depletion between the murine and human systems might be due to the fact that, in the current study, we are looking directly at the effects of IFN treatment and not at IFN induction upon viral infection. As many viruses also have measures to counteract IFN signaling, it may be that previous studies did not observe any differences in the ISG levels in the intestine of IFNAR−/− and IFNLR−/− mice due to specific molecular strategies developed by viruses to counteract the intrinsic innate immune response (Pott and others 2011; Mahlakõiv and others 2015; Nice and others 2015; Lin and others 2016). Indeed, rotavirus, reovirus, and mouse norovirus are all known to display such activities targeting both the IRF3/7 molecules used for type I and type III IFNs induction, as well as the IFN receptors, and their downstream signaling components (Arnold and others 2013a, 2013b; Sen, and others 2017; Stanifer and others 2017). However, unpublished work from our laboratory has shown that human IECs depleted of IFNAR1 have a decreased induction of IFN and ISG transcripts, whereas IFNLR1 depleted cells have an increased induction of IFN and ISG transcripts following reovirus infection. These results suggest that other models are likely to account for the observed differences. Ultimately, this discrepancy could be explained by the inherent difference between murine and human IECs as there is growing evidence that there is major differences in signaling and sensitivity to IFN in a species-specific manner (Mahlakõiv and others 2015; Lin and others 2016; Pervolaraki and others 2017, 2018; Selvakumar and others 2017).
It has been shown that in vivo mice do not respond to type I IFN in their IECs but display basal levels of type III production. As we show that the type III IFN receptor seems to act in a negative manner on type I IFN signaling, maybe the low induction of type I IFN signaling in mice is due to the repression mediated by type III IFN signaling. Interestingly, when intestinal cells from mice are explanted from tissue, they are capable of responding to type I IFN ex vivo (Pott and others 2011; Schwerk and others 2013; Bhushal and others 2017; Selvakumar and others 2017). This suggests that either the signaling is turned down in the context of an animal due to other environmental stimuli or the type I IFN receptor could display a polarized distribution, thereby blocking its signaling in the context of the gut epithelium (Pott and others 2011). Additionally, we have previously demonstrated that human intestinal organoids predominately upregulate type III IFN in response to enteric virus infections; however, they are capable of responding to both type I and type III IFNs (Pervolaraki and others 2017, 2018). While the type I IFN transcript is made, at least in humans, the detection of secreted type I IFN from intestinal cells both in mice and in human organoids has not been shown (Pott and others 2011; Mahlakõiv and others 2015; Pervolaraki and others 2017). This suggests that type I is not made by IECs. Similarly, murine studies support a model where type I IFN emanates from the immune cells located in the lamina propria (Pott and others 2011; Mahlakõiv and others 2015; Lin and others 2016). As we and others have seen that type I IFN signaling acts in a positive manner on type III IFN, one can propose a complex crosstalk between these immune cells and the IECs where type I IFN of hematopoietic origin will reinforce the type III IFN-mediated protection of the mucosal barrier.
Footnotes
Acknowledgments
This work was supported by a research grant from Chica and Heinz Schaller Foundation and Deutsche Forschungsgemeinschaft (DFG) in SFB1129 (Project 14) to S.B. (project No. 240245660), and TRR186 (Project 9; project No. 278001972). This project has received funding from the European Union's Seventh Framework Program under grant agreement number 334336 (FP7-PEOPLE-2012-CIG). M.L.S. is supported by the Olympia Morata Fellowship from Heidelberg University Hospital, the Brigitte-Schlieben Lange Program from the state of Baden Württemberg, Germany, and the Dual Career Support from CellNetworks, Heidelberg, Germany. K.P. is supported with a grant from the A.G. Leventis Foundation, Greece. C.G. was supported by a PhD fellowship from the China Scholarship Council (CSC). We would like to thank Dr. Tim Waterboer and Monika Oppenländer from the German Cancer Research Center (DKFZ) for support with Luminex assay.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.