
Abstract
Chronic hepatitis C virus (HCV) infection accounts for a large proportion of hepatic fibrosis and carcinoma cases observed worldwide. Mechanisms involved in HCV-induced hepatic injury have yet to be fully elucidated. Of particular interest is the capacity of HCV to regulate inflammatory responses. Here, we reveal modulation of cytokine activity by the HCV proteins non-structural protein 3 (NS3), glycoprotein E2, and core protein for their ability to induce chemokine expression in various liver bystander cells. Chemokines sustain chronic liver inflammation and relay multiple fibrogenic effects. CCL2, CCL3, CCL20, CXCL8, and CXCL10 were differentially expressed after treatment of monocytes, fibroblasts, or liver sinusoidal microvascular endothelial cells (LSECs) with HCV proteins. In comparison to NS3 and glycoprotein E2, core protein was a stronger inducer of chemokines in liver bystander cells. Interferon-γ (IFN-γ) and interleukin-1β (IL-1β) synergized with core protein to induce CCL2, CCL20, CXCL8, or CXCL10 in fibroblasts or LSECs. These findings reveal new mechanisms of hepatic injury caused by HCV.
Introduction
Hepatitis C virus (HCV), a single-stranded positive-sense RNA virus, is of the Hepacivirus genus and belongs to the Flaviviridae family. Translation of the HCV genome, which is 9.6 kb in length, gives rise to a polyprotein that is ∼3000 amino acids in length. The generated polyprotein is processed by cellular and viral proteases giving rise to structural (core protein and glycoproteins E1/E2) and nonstructural (NS1 to NS5B) proteins. Once HCV infection is acquired, the majority of patients develop chronic HCV, which is characterized by hepatic fibrosis and hepatocellular carcinoma (Ansaldi and others 2014). Due to the recent development of direct-acting antiviral agents (DAAs), which function by inhibiting viral enzymes, HCV recovery rates have remarkably improved (Omar and others 2018). However, recent studies have reported the emergence of complications associated with the use of DAAs. For instance, resistance-associated substitutions have been identified in treatment-naive patients, particularly in the NS3-, NS5A-, and NS5B-coding regions (Li and others 2017).
Chemokines play a pivotal role in the antiviral response against HCV. CXCR3 ligands, CXCL9 to CXCL11, recruit CXCR3-expressing Th1 cells, cytotoxic T cells, and natural killer (NK) cells to the site of infection. CCR1 and CCR5 expressed on the surface of T cells also play a role in the recruitment of these effector cells (Brass and Brenndörfer 2014). The CCR6 receptor, whose ligands include CCL20, is expressed by B cells, dendritic cells (DCs), NK cells, and T cells (Schutyser and others 2003). CCL20 plays a role in fine-tuning T cell responses by regulating the balance between Th17 and Treg polarization (Ranasinghe and Eri 2018). Furthermore, hepatic recruitment of monocytes relies on the expression of the CCR2 ligand CCL2 (Brass and Brenndörfer 2014). Chemokine expression is primarily regulated by cytokines. Interleukin-1β (IL-1β), a major inflammatory cytokine, upregulates various chemokines such as CCL2, CXCL1, CXCL2, CXCL3, CXCL8, and CXCL10 (Sheng and others 2005; Luo and others 2015). Tumor necrosis factor-α (TNF-α), another principle inflammatory cytokine, induces a similar chemokine profile as IL-1β (Sheng and others 2005; Tourniaire and others 2013). Furthermore, the production of T cell chemoattractants, CXCL9–11, occurs mainly in response to interferon-γ (IFN-γ) (Groom and Luster 2011).
Interestingly, HCV has been found to modulate cytokine expression as a way to evade the immune response. The viral NS3/4A protease complex cleaves mitochondrial antiviral signaling protein (MAVS) and TIR-domain-containing adapter-inducing interferon-β (TRIF), which are signaling molecules downstream of RIG-I-like receptor and Toll-like receptor 3 (TLR3) activation, respectively (Li and others 2005; Ferreira and others 2016). In this manner, the virus inhibits the production of type I and III IFN, thus promoting its own survival. Another HCV protein, the core protein, was shown to inhibit IFN production in human plasmacytoid DCs (pDCs) in response to TLR activation through reduced expression of IFN regulatory factor 7 (IRF7) (Stone and others 2014). Not only does HCV influence cytokine expression but the virus also possesses the capacity to modulate the signaling pathways of cytokines once they are expressed. HCV core protein affects signaling by type I IFN through the Janus kinase/signal transducer and activator of transcription (JAK-STAT) pathway by inducing a dysfunctional increase in nonphosphorylated STAT1 in pDCs (Stone and others 2014). In another study, glycoprotein E2 was found to promote cell survival of Raji cells by overriding FasL-induced cell death through the upregulation of antiapoptotic proteins, Bcl-2 and Bcl-XL (Chen and others 2011).
In this article, we investigate the chemokine inductive profiles of 3 HCV proteins, the core protein, glycoprotein E2, and non-structural protein 3 (NS3), in various liver bystander cells. We observed differential induction of multiple chemokines by HCV proteins. Furthermore, we reveal the capacity of HCV core protein to enhance cytokine activity regarding the induction of chemokine expression in liver bystander cells.
Materials and Methods
Reagents
Recombinant HCV core protein (MBS596029; Sequence ID BAM73001.1), NS3 (MBS596033; Sequence ID AIZ00641.1) and glycoprotein E2 (MBS434003; Sequence ID AAR22408.1) derived from genotype 1b were obtained from MyBiosource (San Diego, CA). The lipopolysaccharide (LPS) content of the recombinant HCV proteins was as low as 0.02 ng/100 μg of core protein, 0.007 ng/100 μg of NS3, and 0.7 ng/100 μg of glycoprotein E2. The maximal level of LPS reached in the highest tested concentrations of the viral proteins did not influence any of the activities reported in this study (data not shown). LPS (O111:B4) derived from Escherichia coli and double-stranded RNA (dsRNA) (P1530) were obtained from Sigma-Aldrich (Overijse, Belgium). Mouse monoclonal IgG2a isotype control (MAB0031) and antibodies against human CCL2 (MAB679 and BAF279), CXCL10 (BAF266), and CXCL8 (MAB208) were obtained from R&D Systems (Minneapolis, MN). Recombinant human CCL3 (270-LD) and recombinant human IFN-γ (285-IF) were obtained from R&D Systems. Capture monoclonal anti-human CXCL10 (500-P93), recombinant human IL-1β (200-01B), and recombinant human TNF-α (300-01A) were obtained from Peprotech (London, UK). Mouse monoclonal anti-TLR2 IgG2a antibody (16-9922-82) was obtained from Thermo Fisher Scientific (Merelbeke, Belgium). TAK-242 (HY-11109) was obtained from MedChemExpress (Monmouth Junction, NJ).
Chemotaxis assay
The migration of peripheral blood mononuclear cells (PBMCs) was determined in Boyden microchamber (Neuro Probe, Gaithersburg, MD) as described (De Buck and others 2013).
Cell cultures
PBMCs and CD14+ monocytes were isolated from human 1-day-old buffy coats, derived from healthy donors (Blood Transfusion Center, Mechelen, Belgium), by density gradient centrifugation and by positive selection (MACS; Miltenyi Biotec, Bergisch Gladbach, Germany) (De Buck and others 2013). Human embryonic skin muscle fibroblasts (E6SM fibroblasts) were grown in minimal essential medium (Lonza, Verviers, Belgium) supplemented with 10% fetal calf serum (FCS). Primary human liver sinusoidal microvascular endothelial cells (LSECs; Cell Systems, Kirkland, WA) were grown in endothelial cell growth medium (EGM; Lonza) supplemented with EGM 2-MV BulletKit (Lonza).
Induction experiments and chemokine quantification
E6SM fibroblasts were seeded in 48-well plates (15,000 cells/cm2, 450 μL/well) in culture medium (Van Damme and others 1989). At 90% confluency, fibroblasts were stimulated with different concentrations of inducers or kept untreated (control) for 72 h at 37°C, 5% CO2. CD14+ monocytes were seeded in 48-well plates (1 × 106 cells/cm2, 450 μL/well) in RPMI 1640 medium supplemented with 0.5% human serum albumin (Belgian Red Cross, Brussels, Belgium). For TLR2 neutralization, CD14+ monocytes were preincubated with an anti-TLR2 antibody for 10 min at 37°C, 5% CO2, before stimulation. Monocytes were stimulated for 24 h.
LSECs were grown to 90% confluency in 48-well plates (15,000 cells/cm2, 500 μL/well), coated with 0.1% gelatin [in phosphate-buffered saline (PBS)] in growth medium, and stimulated for 48 h. For TLR4 neutralization, LSECs were preincubated with a TLR4 antagonist, TAK-242, for 10 min, at 37°C, 5% CO2, before stimulation. Cell supernatants were collected, underwent centrifugation (250 g, 5 min), and were stored at −20°C until further analysis by enzyme-linked immunosorbent assay (ELISA). Human CCL3 and CCL20 were measured with a specific ELISA kit according to the manufacturer's instructions (R&D Systems). Human CCL2, CXCL8, and CXCL10 ELISAs were developed in our laboratory (Schutyser and others 2000; Paul Proost and others 2006).
Flow cytometry
Fibroblasts were seeded in six-well plates at a density of 15,000 cells/cm2. Following a stimulation period of 72 h, the cells were detached by treatment with trypsin for 15 s. Thereafter, trypsin was neutralized and the cells were allowed to recover for a period of 1 h at room temperature in culture medium. To exclude dead cells from the analysis, cells were incubated in Zombie Aqua viability dye (BioLegend, San Diego, CA) in PBS for 15 min at room temperature. To block the Fc receptors, the cells were washed and incubated for 10 min at 4°C with FACS buffer (PBS +2% FCS +2 mM ethylenediamine tetraacetic acid). Afterward, the cells were stained with the anti-human antibodies mentioned hereafter for 30 min at 4°C. Fluorescein isothiocyanate-labeled IL-1R2 antibody (clone REA744) was obtained from Miltenyi Biotech. Phycoerythrin-labeled IL-1R1 (clone 150503) and allophycocyanin-labeled IFN-γR2 (catalogue number FAB773A) were obtained from R&D Systems. Brilliant violet 421 (BV421)-labeled IFN-γR1 (clone GIR-208) was obtained from BD Biosciences (Heidelberg, Germany). The cells were subsequently washed twice and fixed with 0.4% formaldehyde in FACS buffer. Acquisition was carried out using an LSRFortessa X-20 cell analyzer (BD Biosciences) and data analysis was performed using FlowJo software (Tree Star, Ashland, OR).
Statistical analysis
The data did not meet the assumptions of normality; therefore, nonparametric tests were used to perform statistical analysis. The data were initially analyzed using the Kruskal–Wallis test for comparison of multiple groups. Afterward, the Mann–Whitney U test and Wilcoxon signed-rank test were carried out to perform pairwise comparisons using Statistica 13.0 software (StatSoft, Dell, Aliso Viejo, CA). Statistical significance was established at a P-value less than 0.05. The synergy between an HCV protein and IFN-γ or IL-1β in chemokine induction was statistically tested by comparing the chemokine production level in response to combined stimulation, to the sum of the levels measured after treatment with the single stimuli.
Results
HCV proteins fail to induce a direct chemotactic response on PBMCs
We evaluated whether HCV proteins can directly stimulate the migration of human PBMCs in the Boyden microchamber chemotaxis assay (Fig. 1). HCV proteins were found to lack PBMC chemotactic activity. This is evidenced by the low chemotactic indices (CI) observed at the highest tested concentrations of HCV proteins (the core protein at 10 μg/mL; CI 0.4 ± 0.2, NS3 at 10 μg/mL; CI 1.2 ± 0.2, and E2 at 10 μg/mL; CI 0.4 ± 0.1). In contrast, CCL3 provoked pronounced chemoattraction of PBMCs at a concentration of 1 ng/mL (CI 4.5 ± 0.5).

Chemotactic response of PBMCs to HCV proteins. Varying concentrations of HCV core protein (0.4–10 μg/mL), NS3 (0.4–10 μg/mL), glycoprotein E2 (0.4–10 μg/mL), and CCL3 (1 ng/mL) were added to the lower compartment of a Boyden microchamber. Freshly isolated PBMCs were added to the upper compartment and allowed to migrate for 2 h. The chemotactic response is shown as the mean CI+SEM. Results are derived from 3 independent experiments. CI, chemotactic indices; HCV, hepatitis C virus; NS3, non-structural protein 3; PBMC, peripheral blood mononuclear cell; SEM, standard error of the mean.
Distinct chemokine expression in CD14+ monocytes induced by HCV proteins
Dolganiuc and others (2003) have previously shown cytokine induction in monocytes by HCV proteins. To confirm and directly compare the chemokine induction capacity of HCV proteins in CD14+ monocytes, we carried out induction experiments. We observed a relatively potent induction of CCL2 in response to the core protein (4.8 ± 1.9 ng/mL at 10 μg/mL), which is apparent when compared to CCL2 production in response to the bacterial product LPS (5.8 ± 2.2 ng/mL at 5 μg/mL). On the other hand, NS3 (0.8 ± 0.3 ng/mL at 10 μg/mL) did not induce considerable expression of CCL2 in monocytes (Fig. 2A). Furthermore, we also measured CCL3 and CXCL8 expression in CD14+ monocytes in response to stimulation with HCV proteins. The core protein and NS3 induced significant expression of CCL3 (the core protein 10 μg/mL, P-value 0.03, and NS3 10 μg/mL, P-value 0.03). Treatment with 10 μg/mL of the core protein resulted in the expression of 60.5 ± 22.0 ng/mL of CCL3. In contrast, NS3 was less potent; 10 μg/mL induced the expression of 0.3 ± 0.1 ng/mL of CCL3 (Fig. 2B). Furthermore, the core protein induced dose-dependent expression of CXCL8 in CD14+ monocytes (Fig. 2C). Treatment with 10 μg/mL of the core protein resulted in the expression of 153.5 ± 25.4 ng/mL of CXCL8. In contrast, NS3 was less potent; 10 μg/mL triggered the production of 11.4 ± 5.9 ng/mL of CXCL8 (Fig. 2C).
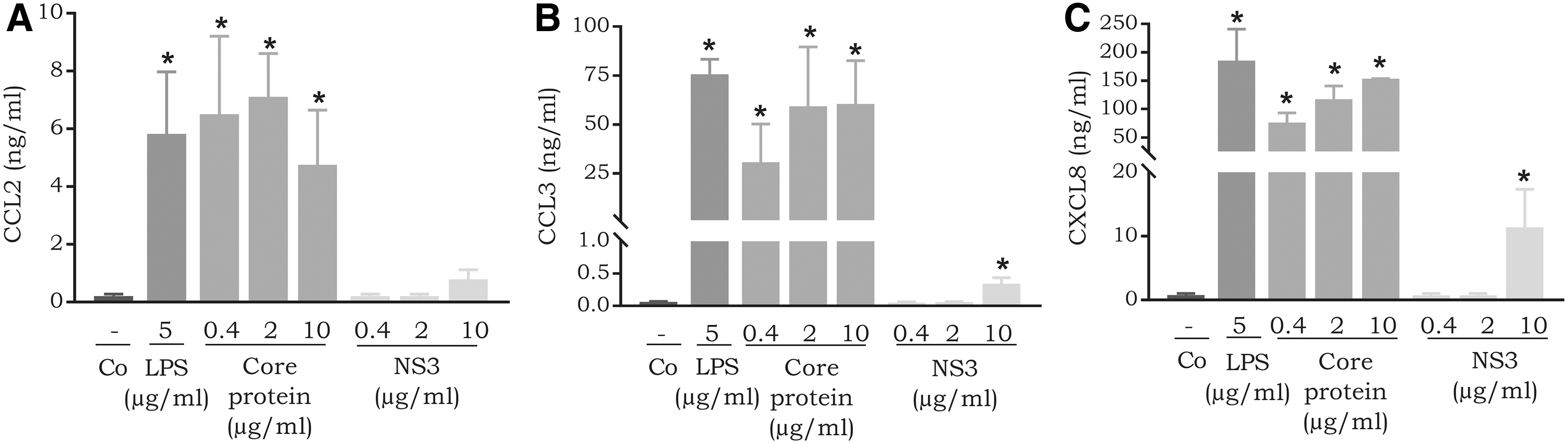
Expression of
Induction of CCL2 and CXCL8 by HCV proteins in fibroblasts and primary human LSECs
In addition to CD14+ monocytes, we analyzed CCL2 and CXCL8 expression in response to HCV proteins in fibroblasts and LSECs (Fig. 3). Compared to the potent chemokine inducers IL-1β, TNF-α, and dsRNA, the viral proteins were not powerful CXCL8 inducers in fibroblasts. However, at 5 μg/mL, glycoprotein E2 was found to significantly induce low quantities of CXCL8 (1.2 ± 0.4 ng/mL; P-value 0.02) in fibroblasts (Fig. 3B). Similarly, the viral proteins were very weak inducers of CCL2 in fibroblasts. However, stimulation with the core protein resulted in a dose-dependent increase in CCL2 expression (Fig. 3A; Mann–Whitney U test, P-value 0.09; Wilcoxon signed-rank test, P-value 0.04).
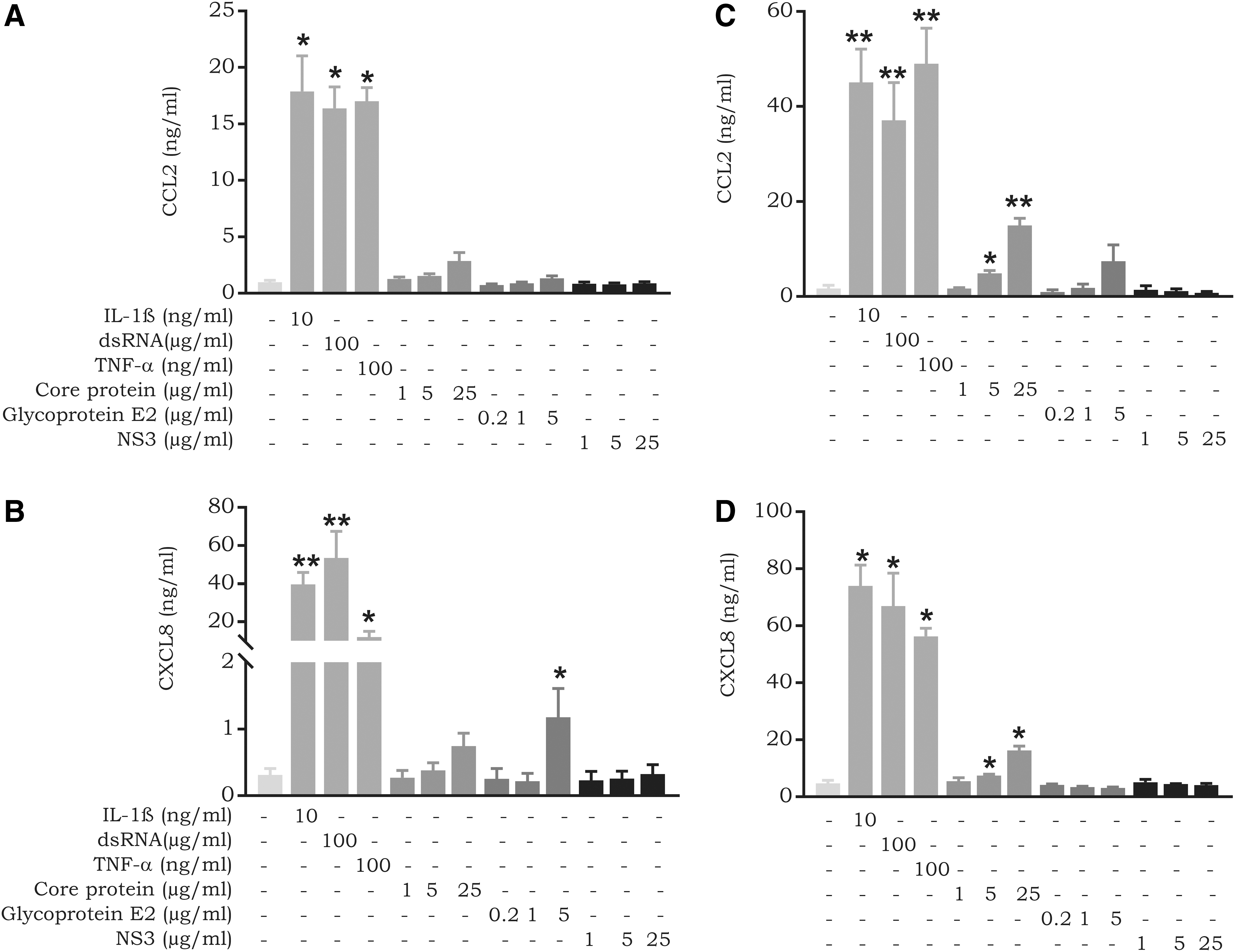
Expression of CCL2 and CXCL8 by fibroblasts and LSECs in response to HCV proteins.
In contrast, the core protein, although weaker than IL-1β, TNF-α and dsRNA, was a significant inducer of CCL2 (15.0 ± 1.5 ng/mL; P-value 0.005) and CXCL8 (16.3 ± 1.4 ng/mL; P-value 0.01) in LSECs at 25 μg/mL (Fig. 3C, D). Besides CXCL8 and CCL2, we measured the expression of the CXCR3 ligand CXCL10, but we observed no expression in fibroblasts or LSECs after treatment with the viral proteins (data not shown).
Synergy between HCV core protein and IFN-γ to produce CCL2, CXCL8, and CXCL10 in LSECs
The NS5a protein has been previously shown to synergize with a cytokine mixture (IL-1β, TNF-α, and IFN-γ) to induce chemokine expression in the human Chang liver cell line (Apolinario and others 2005). To determine whether HCV proteins influence chemokine expression in response to IFN-γ, we carried out induction experiments with combinations of IFN-γ and HCV proteins (the core protein, NS3, and glycoprotein E2) on LSECs. Interestingly, a combination of the core protein with IFN-γ induced a dose-dependent enhancement of CCL2, CXCL8, and CXCL10 expression (Fig. 4). Being a principal inducer of CXCR3 ligands, IFN-γ alone stimulated significant expression of CXCL10 (7.0 ± 1.8 ng/mL at 200 ng/mL; P-value 0.005), but not of CCL2 or CXCL8 (Fig. 4). Remarkably, the production of CXCL10 increased more than 10-fold (up to 135.4 ± 52.0 ng/mL) when the core protein was combined with IFN-γ. For CXCL8, a production level up to 33.0 ± 4.6 ng/mL was reached when 25 μg/mL of the core protein was combined with 200 ng/mL of IFN-γ, compared to 16.3 ± 1.3 ng/mL of CXCL8 obtained following treatment with 25 μg/mL of the core protein alone. The levels obtained for CCL2 with these stimuli were very comparable. NS3 and glycoprotein E2 did not influence the inductive capacity of IFN-γ in LSECs (data not shown).
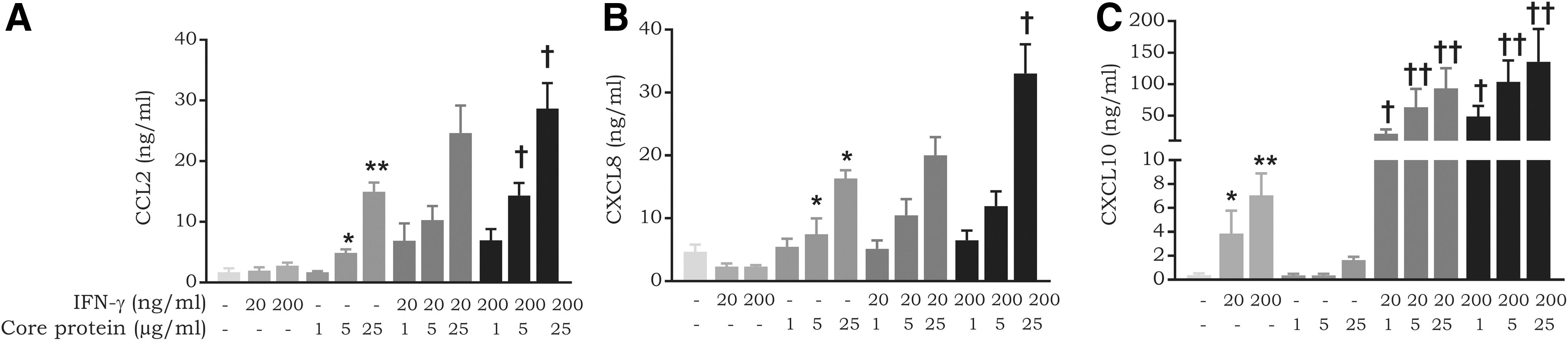
Synergy between the core protein and IFN-γ in chemokine expression in LSECs. LSECs were induced with combinations of varying concentrations of IFN-γ (20–200 ng/mL) and core protein (1–25 μg/mL). Following an incubation period of 72 h, cell supernatants were collected and
Synergy between HCV core protein and IFN-γ to produce CCL2, CCL20, CXCL8, and CXCL10 in fibroblasts
The occurrence of synergy between IFN-γ and HCV proteins was also determined in fibroblasts (Fig. 5). In contrast to what was observed on LSECs, we did not detect the expression of CXCL10 in response to IFN-γ alone in fibroblasts. Nevertheless, we observed synergy between IFN-γ and the core protein in the expression of CXCL10 (∼10-fold upregulation). Furthermore, synergy was also exceedingly pronounced for induction of CCL2, CCL20, and CXCL8 in response to combinations of IFN-γ and the core protein. Nevertheless, the maximal production levels reached were lower than those observed in LSECs (Fig. 4). NS3 and glycoprotein E2 did not influence chemokine expression in response to IFN-γ in fibroblasts (data not shown).
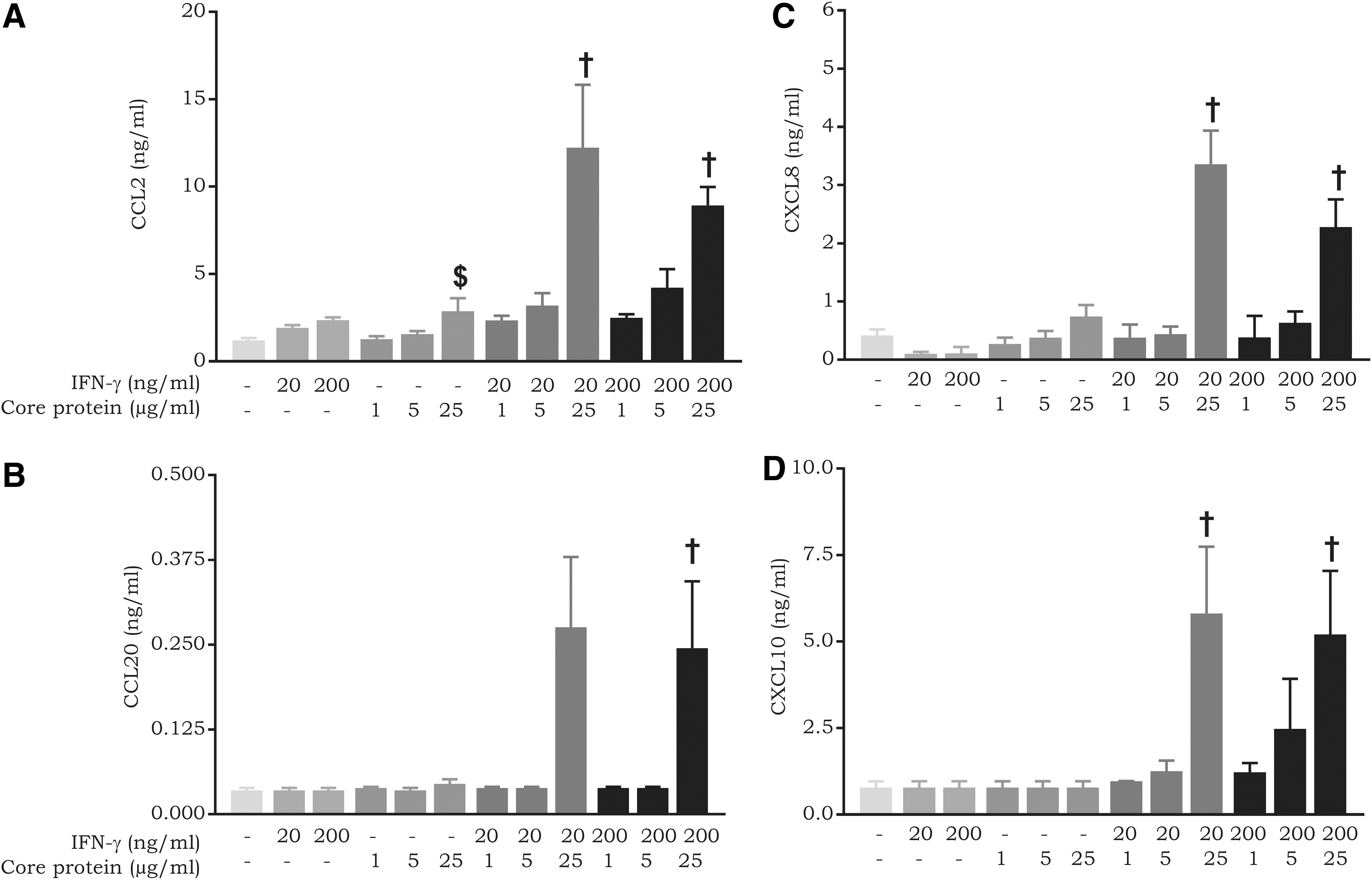
Synergy between the core protein and IFN-γ in chemokine expression in fibroblasts. Fibroblasts were induced with combinations of varying concentrations of IFN-γ (20–200 ng/mL) and core protein (1–25 μg/mL). Following an incubation period of 72 h, cell supernatants were collected and
Core protein and IL-1β synergistically induce CCL20 expression in fibroblasts
To determine whether the HCV proteins influence chemokine expression in response to IL-1β, we carried out induction experiments with combinations of IL-1β and HCV proteins on fibroblasts and LSECs. A dose-dependent synergistic effect was observed between the core protein and IL-1β in the production of CCL20 by fibroblasts (Fig. 6B). For CCL20, statistical significance was reached, but for CXCL8, synergy was not obtained in all experiments. Rather, an overall additive effect was observed. This is possibly due to the fact that IL-1β is a strong CXCL8 inducer in fibroblasts (Fig. 6A). The production levels reached were higher when the core protein was combined with IL-1β than with IFN-γ; for CCL20, the maximal production level was 14.0 ± 0.1 ng/mL after stimulation with the core protein plus IL-1β in fibroblasts, versus 0.3 ± 0.1 ng/mL after stimulation with the core protein plus IFN-γ in LSECs. On the contrary, no synergy was observed between the viral proteins and IL-1β in LSECs. Furthermore, glycoprotein E2 and NS3 did not influence IL-1β activity in fibroblasts and LSECs (data not shown).

Core protein and IL-1β synergistically induce CCL20 expression in fibroblasts. Fibroblasts were induced with combinations of varying concentrations of IL-1β (0.1–1 ng/mL) and core protein (5–25 μg/mL). Following an incubation period of 72 h, cell supernatants were collected.
HCV proteins utilize TLRs during chemokine induction
Previous studies have suggested that HCV proteins utilize TLRs for cytokine induction in various cell types (Dolganiuc and others 2004; Hosomura and others 2011; Swaminathan and others 2014; Rajalakshmy and others 2015). To determine whether TLR2 is activated by the core protein and NS3 in CD14+ monocytes, receptor neutralization was carried out using an anti-TLR2 antibody. Although not statistically significant, TLR2 neutralization decreased the CXCL8 inductive capacity of NS3 by 23% (217.9 ± 94.0 ng/mL versus 165.0 ± 69.3 ng/mL; P-value 0.45) (Fig. 7A). On the other hand, TLR2 neutralization did not influence chemokine expression in response to the core protein. However, the core protein has also been described to utilize TLR4 (Uraki and others 2015). Therefore, LSECs were stimulated with either core protein alone or in combination with IFN-γ in the presence of the TLR4 antagonist TAK-242. TAK-242 dramatically inhibited the CXCL8-inducing capacity of the core protein (6.2 ± 0.6 ng/mL versus 0.6 ± 0.1 ng/mL; P-value <0.0001). Furthermore, synergy with IFN-γ was completely inhibited following treatment with TAK-242 (97% inhibition; P-value 0.0002) (Fig. 7B).
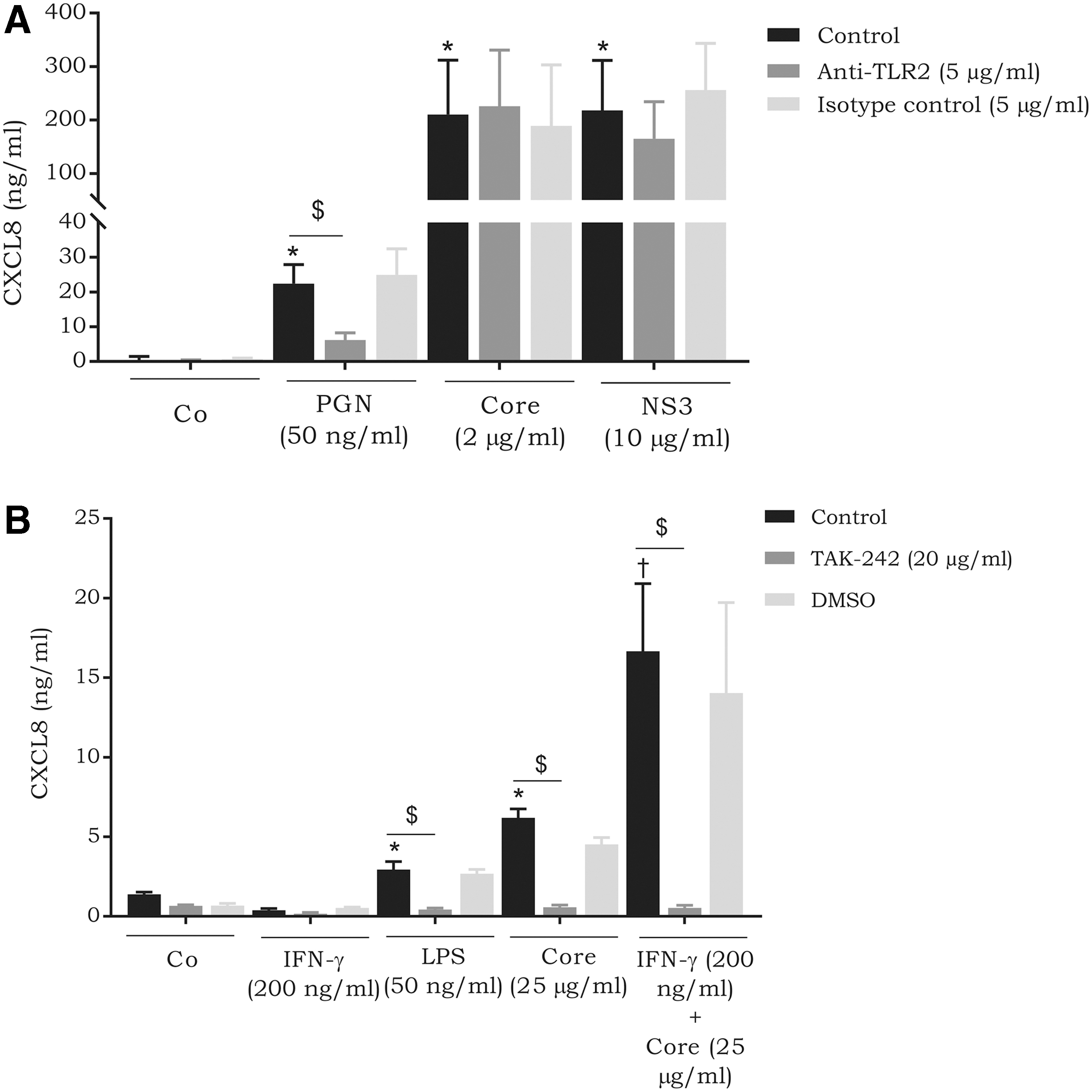
The effect of TLR neutralization on the chemokine-inducing capacity of NS3 and HCV core protein.
HCV core protein does not regulate the expression of IFN-γ and IL-1β receptors in fibroblasts
To reveal the molecular mechanism behind the synergy between the HCV core protein and IL-1β or IFN-γ, we verified cytokine receptor expression, as Hosui and others (2003) demonstrated upregulation of IFN-γR2 on murine liver cells expressing HCV core protein. To determine whether synergy in chemokine induction (Figs. 5 and 6) between the core protein and IFN-γ or IL-1β is in fact due to receptor modulation by the core protein, we assessed the expression levels of IL-1R1, IL-1R2, IFN-γR1, and IFN-γR2 on fibroblasts using flow cytometry. No significant upregulation of IL-1R1, IL-1R2, IFN-γR1, and IFN-γR2 was observed on fibroblasts following stimulation with 25 μg/mL of the core protein (Fig. 8).
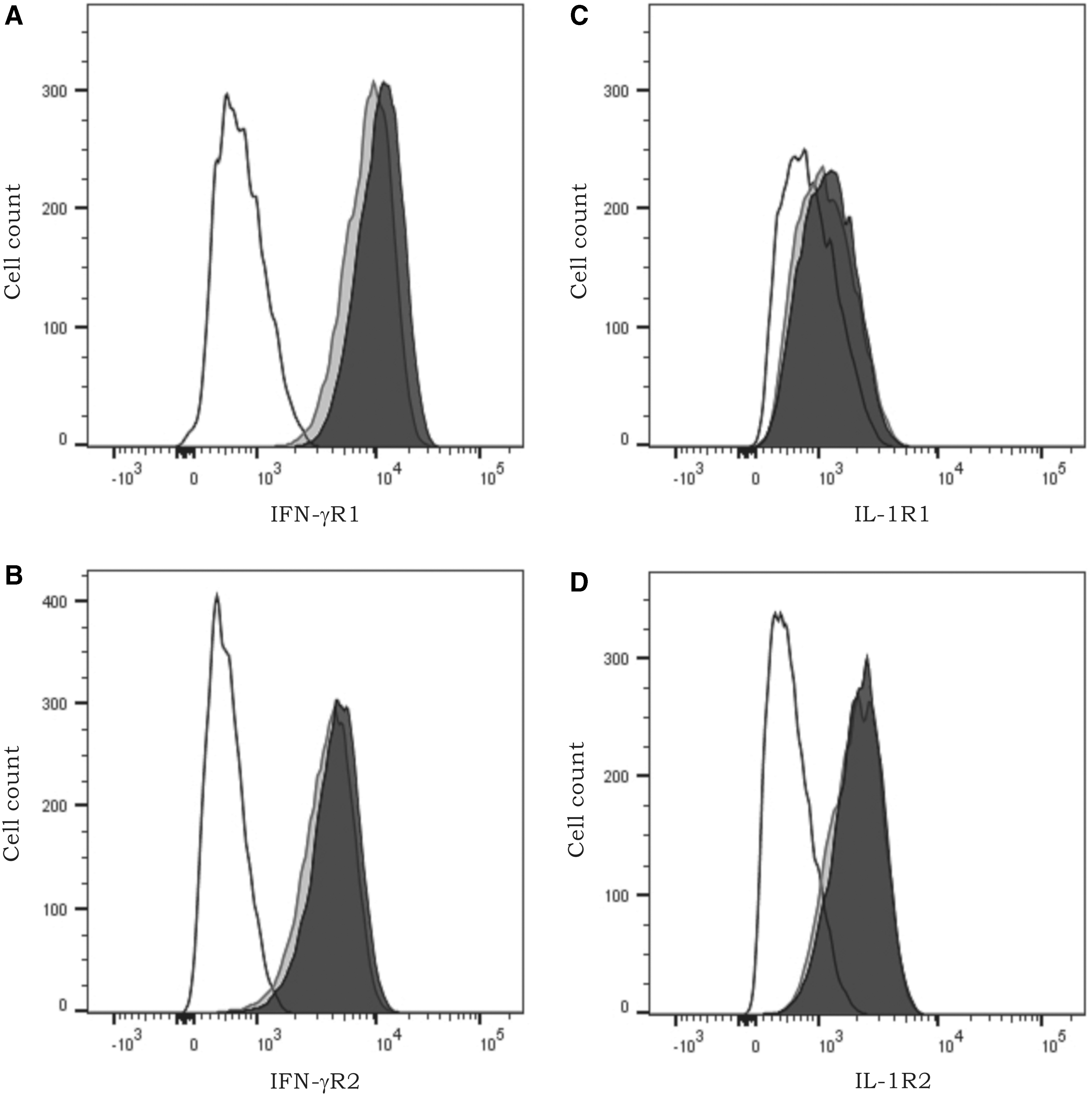
The effect of HCV core protein on the expression of IFN-γ and IL-1β receptors on fibroblasts. Fibroblasts were treated with 25 μg/mL of core protein or left untreated. Following an incubation period of 72 h, the expression of
Discussion
In this article, we demonstrate varying chemokine induction profiles of HCV proteins on distinct cell types. Certainly, such distinct chemokine expression is to be anticipated among different cell types due to the variable expression of receptors for these viral proteins. Various receptors have been described to be utilized by HCV proteins. However, the majority of studies have attributed the cellular effects of most HCV proteins to TLR activation. The core protein has been reported to utilize TLR2, in addition to TLR1 and TLR6 as co-receptors (Dolganiuc and others 2004; Chang and others 2007). To a lesser extent, the core protein has also been described to interact with TLR4 (Uraki and others 2015). In another study, the core protein was described as a globular C1q receptor ligand (Song and others 2016). Similarly, NS3 has been demonstrated to activate TLR2 and TLR4 (Hosomura and others 2011; Rajalakshmy and others 2015). In line with this, we observed a reduced chemokine inductive capacity for the core protein in LSECs in the presence of a TLR4 antagonist. Similarly, the synergy between the core protein and IFN-γ for chemokine induction in LSECs was drastically diminished following TLR4 antagonization. These data suggest that the core protein and IFN-γ activate their individual receptors leading to amplified downstream signaling, which is translated into enhanced chemokine expression. Chemokine induction by HCV core protein in CD14+ monocytes was independent of TLR2; in contrast, the CXCL8 production in response to NS3 stimulation was somewhat reduced in the presence of a neutralizing anti-TLR2 antibody.
In this study, we investigated the cytokine induction spectrum of 3 different HCV proteins. One could question the relevance of these recombinantly expressed viral proteins as inducers. However, as an envelope protein, E2 is certainly visible to extracellular sensors (Dubuisson 2007). In addition, circulating core protein has been detected in serum from patients and has been used in several studies to investigate stimulation of cytokine production by HCV (Tanaka and others 1996; McLauchlan 2000; Cividini and others 2003; Dolganiuc and others 2003, 2004). Finally, HCV has been demonstrated to induce a specific form of programmed cell death, namely, pyroptosis, which is an inflammatory form of cell death, whereby the infected cell lyses and releases its contents into the extracellular environment (Kofahi and others 2016). Therefore, pyroptosis leads to the extracellular occurrence of otherwise intracellular HCV proteins.
We observed differential expression of CCL2 and CXCL8 in CD14+ monocytes, fibroblasts, and LSECs in response to the HCV core protein, glycoprotein E2, and NS3. On the other hand, we did not observe expression of CCL20 and CXCL10 after stimulation with one of these viral proteins as a single stimulus. CXCL8 is probably the most frequently reported chemokine to be expressed in response to HCV proteins. Glycoprotein E2 was found to induce the expression of CXCL8 in primary human thyroid cells and human umbilical vein endothelial cells (Urbaczek and others 2014; Hammerstad and others 2017). In addition, NS5a induces the expression of CXCL8 in monocyte-derived DCs (Wertheimer and others 2007). In contrast to CXCL8, CCL2 expression in response to HCV proteins has only been reported in human hepatic stellate cells (HSCs) (Bataller Ramó and others, 2004). Previous studies have documented the expression of additional chemokines in response to HCV proteins. Expression of CCL5 mRNA in the Chang liver cell line was shown to occur in response to constitutively expressed HCV core protein (Ruggieri and others 2007). Brenndörfer and others (2012) demonstrated an increase in hepatic expression of murine CCL17 and CCL22 in response to the transgenic expression of the HCV protease complex NS3/4A.
Not only do HCV products directly induce the expression of chemokines but they also possess the capacity to synergize with cytokines to enhance said chemokine expression. Helbig and others (2009) documented an increase in CXCL11 mRNA expression under IFN-γ plus TNF-α stimulation in HCV-infected Huh-7 cells. Further experiments revealed that synthetic dsRNA mediated a synergistic effect with IFN-γ and TNF-α by enhancing signaling through the RIG-I/IRF3 pathway. Apolinario and others (2005) previously showed that the core protein and NS5a could synergize with a cytokine mixture, consisting of IL-1β, IFN-γ, and TNF-α, to enhance the expression of T cell-specific chemokines in Chang liver cells. We demonstrated, for the first time, a several-fold increase in chemokine expression in response to IFN-γ, in the presence of the core protein, in fibroblasts and LSECs. It is worth noting the magnitude of this increment, particularly in the case of LSECs. In those cells, we observed a 10-, 13-, and 19-fold increase in CCL2, CXCL8, and CXCL10 expression, respectively. Although not to the same intensity as that seen with IFN-γ, we detected direct synergy between the core protein and IL-1β in the induction of CCL20 expression in fibroblasts.
The observed synergy promotes speculation on how the virus may benefit from such a drastic increase in chemokine expression. A plausible explanation is that the virus may have evolved to prolong its host's survival by reducing its own pathogenicity, thus ensuring its host remains in a healthy condition for a longer duration of time. Initially, the presence of these chemokines would serve to be beneficial, as it would contribute to viral elimination through the recruitment of various leukocytes. However, persistent exposure to certain chemokines may result in hepatic injury. A hallmark of chronic HCV infection is the inflammatory damage that is inflicted upon the liver, thus leading to the development of fibrosis and cirrhosis. Of note, the concentrations we measured in the culture supernatants are biologically relevant. Indeed, the minimal effective concentration of these chemokines (CCL2, CCL3, CCL20, CXCL8, and CXCL10) in chemotaxis assays falls within the range of 1–10 ng/mL (Van Damme and others 1989, 1992; Proost and others 2001; Schutyser and others 2003).
The occurrence of synergy between HCV proteins and cytokines in vivo and the implications it would bring about on disease progression is intriguing. LSECs line the hepatic sinusoidal space and are thus within close proximity of hepatocytes and HSCs. As a result, upregulated chemokine expression is to be expected within the hepatic environment. Upon hepatic injury, HSCs are activated and differentiate into proliferative myofibroblasts that drive fibrosis (Tacke and Weiskirchen 2012). CCL2 contributes to the activation of HSCs through multiple pathways. CCL2 brings about a fibrotic state by inducing angiogenesis in the portal veins. This angiogenic effect is moderated by CCL2-mediated macrophage infiltration (Ehling and others 2014). Subsets of murine macrophages have been defined and fall within at least 2 categories, LY6Chi and LY6Clow, which display opposing effects in terms of their fibrogenic capacities. The fibrogenic subset, LY6Chi macrophages, activates HSCs through the expression of various cytokines, including IL-1β, CCL2, CCL3, CCL5, and platelet-derived growth factor (Tsuchida and Friedman 2017). Furthermore, the role of the CCL2 receptor, CCR2, has been established in the process of fibrosis. Murine models of hepatic inflammation demonstrated the upregulation of CCR2 in HSCs. Furthermore, upregulation of CCR2 corresponded to several fibrogenic findings in HSCs such as enhanced migration and upregulation of fibrogenic mediators such as reactive oxygen species (Seki and others 2009).
CXCL8 is another chemokine found to promote fibrosis. Upregulated CXCL8 is observed in patients with chronic liver disease and correlates with activated HSCs and enhanced expression of α-smooth muscle actin (Clément and others 2010; Zimmermann and others 2011). Furthermore, NK cells have been described to relay an antifibrotic effect by induced apoptosis of HSCs. Hintermann and others (2010) revealed an inhibitory role of CXCL10 on the antifibrotic effect of NK cells. CCL20, whose expression is upregulated in other viral infections, was recently found to display fibrogenic effects. Affò and others (2014) demonstrated a chemoattractant effect of HSCs toward CCL20 by ERK signaling. Furthermore, silencing of CCL20 resulted in the downregulation of LPS-induced liver damage, thus suggesting that CCL20 may play a role in hepatic fibrosis. The presence of chemokines, such as CCL2 and CCL3, in the hepatic vicinity would induce the recruitment of monocytes that would further escalate the inflammatory response by their own chemokine expression, thus creating an amplification feedback loop. The potential pathogenic activities of chemokines in HCV-induced liver disease are summarized in (Fig. 9). Taken together, our data suggest that LSECs might promote HCV-induced hepatic injury through the production of chemokines that recruit leukocytes and activate HSCs in both a direct and indirect manner.

An overview of the inflammatory damage inflicted by expressed chemokines. HSCs, hepatic stellate cells. Color images are available online.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Research Foundation of Flanders (FWO-Vlaanderen) and C1 funding [C1 Project number C16/17/010] of the KU Leuven. MG is a research expert funded by the Rega foundation. MDB is a postdoctoral research fellow of the FWO-Vlaanderen.