
Abstract
Tumor necrosis factor alpha (TNF-α) plays a paramount role in homeostasis by inducing tumor cytotoxicity and activating immune system. The signaling complexes formed by TNFR1 to activate JNK, p38, and nuclear factor-kappa B pathways and to subsequently induce apoptosis and necroptosis are well known. However, this “canonical TNF-α signaling” does not explain how ERK, AKT, and STAT3 can be activated by TNF-α. In addition, little to nothing is known about negative regulation of TNFR1 signaling. Because cyclic AMP-activated kinase (PKA) shows anti-TNF and anti-inflammatory activities, we postulated that PKA might affect TNF-α signaling by directly phosphorylating TNFR1. In line with this, we identified 2 putative PKA-phosphorylation motifs RRRT411 and REAT417 within the death domain of TNFR1, and investigated whether “canonical” and “noncanonical” TNFR1 signaling is regulated by modifications of T411 and T417. In this study, we demonstrate for the first time that PKA directly binds to and phosphorylates TNFR1 after TNF-α stimulation. To further support our hypothesis, we generated alanine and phosphomimetic (aspartic acid) mutants of TNFR1 at positions T411 and T417, ectopically expressed these mutants, and determined their influence on TNF-α-induced activations of ERKs, AKT, STAT3, p38α, and JNK1/2. Our results clearly showed that phosphomimetic mutants significantly suppressed and alanine mutants augmented TNF-α-induced phosphorylations of ERKs, AKT, Stat3, p38α, and JNKs. These findings strongly suggest that PKA-mediated phosphorylation of T411 and T417 of TNFR1 interferes with both “canonical” and “noncanonical” TNF-α signaling.
Introduction
Tumor necrosis factor alpha (TNF-α) is a multifunctional cytokine, which plays indispensable role in activation of immune system, inflammation, modulation of growth, differentiation, and tumor cytotoxicity (Sedger and McDermott 2014; Kalliolias and Ivashkiv 2016). TNF-α elicits its biological effects by binding to 2 different receptors, TNFR1 and TNFR2. TNFR1 is a 55 kDa transmembrane protein with death domain (DD) on its cytoplasmic domain (aa 356–441) and interacts with other DD bearing proteins such as TRADD and RIP1 (Hsu and others 1995, 1996a, 1996b). Although binding of TRADD to TNFR1 recruits the Fas-associated DD and procaspase-8 into a complex that initiates apoptotic caspase cascade, binding of TRADD to RIP1 and TNF-receptor-associated factor 2 (TRAF2) induces formation of complex-I. Complex-I is composed of RIP1 (receptor-interacting serine-threonine kinase 1), TRAF2, cIAP1/2 (cellular inhibitor of apoptosis protein 1/2), LUBAC (linear ubiquitin chain assembly complex), and TAK1 (transforming growth factor-activated kinase-1) (Rothe and others 1995; Shu and others 1996; Park and others 2004; Ea and others 2006; Zheng and others 2006; Jackson-Bernitsas and others 2007; Bertrand and others 2008; Varfolomeev and others 2008; Haas and others 2009; Tokunaga and others 2009; Gerlach and others 2011). According to the most widely accepted “canonical TNFR1 signaling” model, once this ubiquitin network is established on TNFR1, TRAF2, cIAP1, and cIAP2 introduce Lys63-linked ubiquitination on RIP1, NEMO, and TAK1, then LUBAC complex adds Met1-linked ubiquitin chains, resulting in stabilization of complex-I. In this activated complex, TAK1 induces activation of mitogen-activated protein kinase kinases, MKK3/6, MKK4/7, and IKK complex, resulting in activation of p38, JNKs, and nuclear factor-kappa B (NF-kB) pathway (Wang and others 2001).
The cyclic AMP-activated kinase (PKA) is known for its anti-inflammatory and anti-TNF activities for a long time. Several studies made correlative association between PKA activation and inhibition of TNF-α synthesis or TNF-α signaling. Among these studies, Wall and others (2009) have shown that prostaglandin E2 (PGE2), produced by activated macrophages, functions in an autocrine manner and plays significant role tocoordinate the immune response, and reverse lipopolisaccharide-dependent Toll-like receptor-induced activation of macrophages by inhibiting TNF-α synthesis, and this activity of PGE2 was shown to be dependent on cAMP-dependent protein kinase (PKA) (Wall and others 2009). In line with this, Deere and others (2008) have also shown that synthesis of TNF-α in activated macrophages is inhibited by activation of PKA. PKA-mediated downregulation of TNF-α synthesis or abrogation TNF-α-induced gene induction has very strong biological consequences and explains mode of action of several anti-inflammatory drugs. Among these, the immunoregulatory and anti-inflammatory function of apremilast and methotrexate were shown to be dependent on production of cAMP and activation of PKA, Epac1, and Epac2 (Perez-Aso and others 2015), and adiponectin-dependent abrogation of TNF-α-mediated induction of NF-kB and plasminogen activator inhibitor-1 (PAI1) is mediated by activation of PKA by adiponectin (Chen and others 2017).
Although the activation of “canonical TNF-α signaling complex” can explain TNF-α-induced activations of p38, JNKs, and NF-kB, the mechanism of TNF-α-induced bindings and activations of Erk, Akt, Stat3, Stat5, and PI3Kp85α is elusive and cannot be explained with this model. Therefore, we believed and identified complementary TNF-α-activated pathway and we called this “noncanonical TNF-α signaling pathway.” This pathway is initiated by tyrosine phosphorylation of TNFR1 at Y360 and Y401 of TNFR1 and these phosphorylations mediate TNF-α-induced activations/phosphorylations of Erk, Akt, Stat3, Stat5, and PI3Kp85α. Moreover, to elucidate the molecular basis of PKA-mediated downregulation of TNF-α signaling, we searched and identified 2 putative PKA phosphorylation sites at T411 and T417 positions of TNFR1 and hypothesized that PKA-mediated negative regulation of TNF-α signaling may be mediated by direct modulation of TNFR1 by PKA at these sites. In line with this, for the first time, we showed that PKA binds to and phosphorylates TNFR1 after TNF-α stimulation. To further support our hypothesis, we generated alanine and aspartic acid mutants of TNFR1 at T411 and T417 positions, ectopically expressed these mutants, and determined the levels of TNF-α-induced activation of ERKs, AKT, Stat3, p38α, and JNK1/2. Our results clearly showed that although phosphomimetic aspartic acid mutants significantly suppressed, alanine mutants augmented TNF-α-induced phosphorylations of ERKs, AKT, Stat3, p38α, and JNKs. These findings strongly suggest that PKA-mediated phosphorylation of T411 and T417 of TNFR1 interferes with TNF-α signaling and elucidates the molecular basis for anti-inflammatory and anti-TNF activity of cAMP and activation of PKA.
Materials and Methods
Antibodies and reagents
Recombinant human TNF-α was purchased from Sigma (H8916), protein A/G-Agarose beads (sc-2003) and monoclonal primary antibodies for His-probe (sc-803), TNFR1 (sc-8436), pERK (sc-7383), ERK (sc-94), pY20 (sc-508 and sc-508 AC), Grb2 (sc-255), c-Src (sc-8056) and GAPDH (sc-47724), and standard rabbit and mouse immunoglobulin G (IgG) for immunoprecipitations (sc-2027 and sc-2025) were from Santa Cruz Biotechnology. Monoclonal primary antibodies for pAkt (9271, 9275), Akt (9272), and p85α (13666S) were from Cell Signaling (NEB) and horseradish peroxidase (HRP)-conjugated secondary antibodies to mouse and rabbit IgG was from KPL (474–1806 and 474–1506).
Plasmids and mutagenesis
Following total RNA isolation from 293T cells using High Pure RNA isolation kit (1828665001; Roche), cDNA library was established with the aid of cDNA Synthesis System (11117831001; Roche) according to the manufacturer's instructions. TNFR1 cDNA was amplified from the cDNA library using 5′-cgcggatcc gccgccatgggcctctccaccgtgcctgacctg-3′ forward and 5′-cgcggatcctctgag aagactgggcgcgggcgggagggcggc-3′ reverse primers with Qiagen Taq DNA polymerase. The reaction conditions were as follows: 94°C 5 min predenaturation; 35 cycles composed of 94°C 45 s, 60°C 1 min, 72°C 3 min, and 72°C 5 min final extension. The obtained PCR product and pcDNA3.1a plasmid backbone were digested with BamH1 restriction (NEB). Digested plasmid backbone and TNFR1 construct was ligated using T4 DNA ligase (NEB) according to the manufacturer's instructions. Sense orientation of cloned plasmids was verified by EcoRI digestion (NEB), Sanger sequencing, and transfection into HEK293T cells. Site-directed mutagenesis was performed by Pfu polymerase (Agilent Stratagene) reaction and plasmid sequences were verified by Sanger sequencing using Applied Biosystems 3130XL with 5′-gcgtttaaacgggccctctagactc-3′ forward and 5′-gtcgacggcgctattcagatcctc-3′ reverse primers.
Cell culture, transfections, and treatments
HEK293T cells obtained from ATCC were grown in high glucose Dulbecco's Modified Eagle's Medium (DMEM) containing stable
Immunoprecipitation and Western blot
Protein concentrations of whole cell lysates obtained in lysis buffer (20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1.2% Triton X-100, 1 mM EGTA, 1 mM EDTA, 1 mM PMSF, 0.15 U/mL aprotinin, 10 g/mL leupeptin, 10 g/mL pepstatin A, and 1 mM Na3VO4) were determined using Bradford method. Before immunoprecipitations, lysates were precleared with 50 μL protein A/G-Agarose beads. To precipitate the protein complexes, 2 μg primary antibody was added per 1 mg protein lysate, shaken for 2 h in +4°C, then protein A/G-agarose beads were added and shaken overnight in 4°C. Beads pelleted by centrifugation at 6000 g for 1 min were washed in cold lysis buffer at least 3 times and proteins were eluted in sodium dodecyl sulfate (SDS) sample buffer containing β-mercaptoethanol by boiling for 5 min. Equal amounts of protein were fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 10% polyacrylamide gels and transferred overnight to Immobilion-P polyvinylidene difluoride (PVDF) membranes. Blots were probed with primary antibodies and corresponding HRP-conjugated secondary antibodies, and proteins were detected using Clarity ECL Western blotting substrate (1705061; Bio-Rad).
Statistical analysis
Western blot image intensities were measured by ImageJ software. For immunoprecipitation studies, band intensities were proportioned to IgG band intensities. For protein phosphorylation studies, intensities of protein phospho-forms were proportioned to intensities of total protein bands. Graphs were drawn with GraphPad Prism software and data were expressed as mean ± standard error of the mean. For statistical analysis, Mann–Whitney U-test was performed using the same software.
Results
TNF-α-induced tyrosine phosphorylation of TNFR1 is inhibited by PKA-mediated phosphorylation of TNFR1
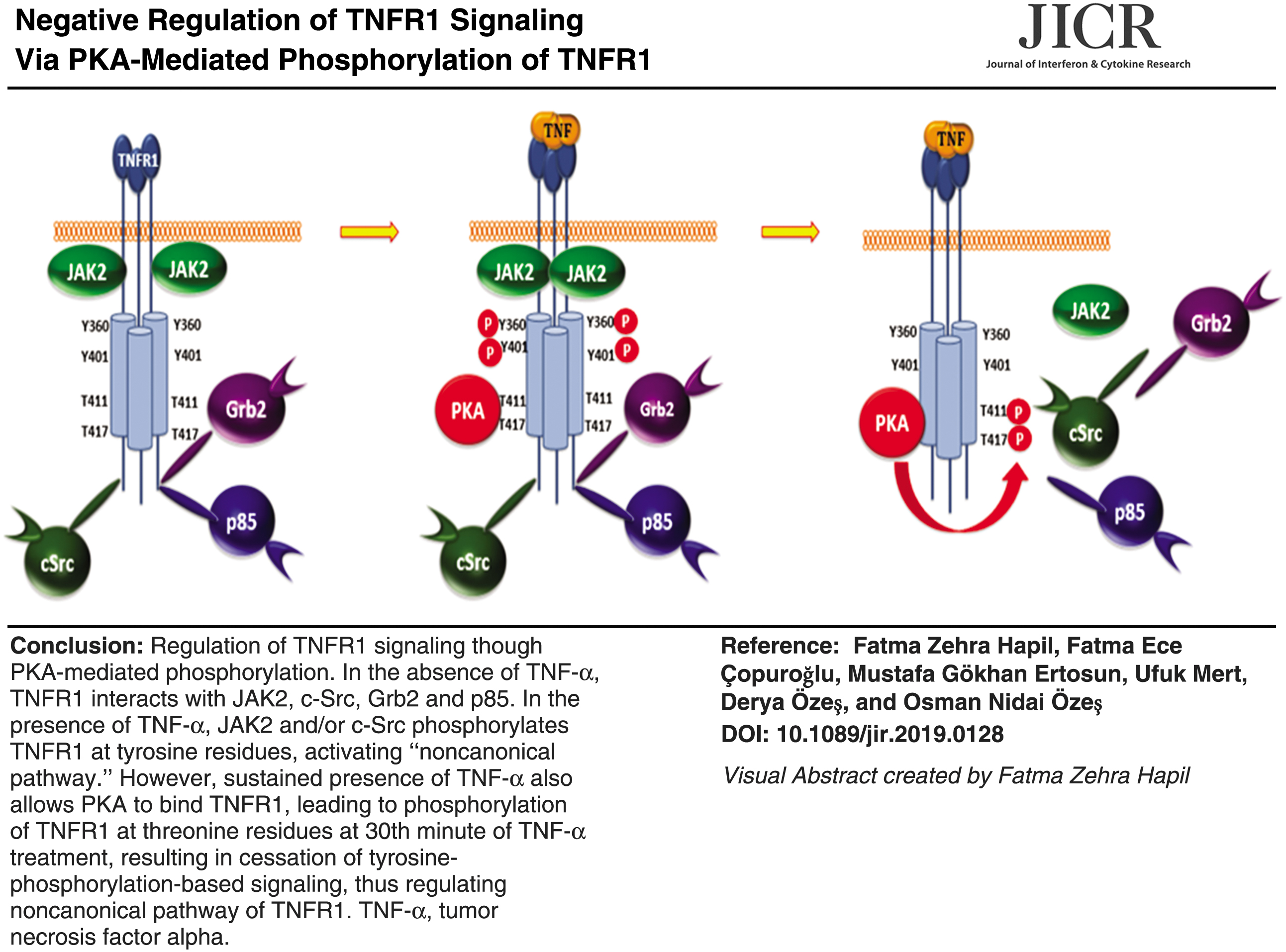
When we analyzed the amino acid sequence of human TNFR1 (NCBI accession number: AAA61201.1), we found 2 tyrosine and 2 PKA phosphorylation site motifs in the DD of TNFR1 (Fig. 1A) and searched PhosphositePlus database and found that Y360, Y401, T411, and T417 were reported to be phosphorylated. Then, we postulated that although tyrosine phosphorylations may play positive role in TNF-α signaling, PKA-mediated phosphorylations of T411 and T417 may have an negative regulatory role in TNFR1 tyrosine phosphorylation. Indeed, we have shown that TNFR1 is phosphorylated at tyrosine residues after TNF-α stimulation in a time-dependent manner, reached the maximum at 30th minute, and these phosphorylations were carried out by JAK2 and c-SRC (Hapil and others, submitted for publication). To support our hypothesis, we pretreated 293T cells with the PKA activator Forskolin for 1 h, then stimulated cells with TNF-α for 30 min, and determined the level of tyrosine phosphorylation of TNFR1. As given in Fig. 1B, TNF-α stimulated tyrosine phosphorylation of TNFR1, and this was profoundly inhibited by Forskolin. In line with this observation, treatment of cells with PKA inhibitor (PKI) potentiated TNFR1 tyrosine phosphorylation (Fig. 1C), suggesting a negative role of PKA on TNFR1 tyrosine phosphorylation. To examine the possibility of a physical interaction between TNFR1 and PKAcα, we immunoprecipitated TNFR1 and PKAcα from TNFR1-transfected 293T cells, and blotted with reciprocal antibodies. As given in Fig. 1D and E, TNF-α promoted interaction of PKA with TNFR1. Because TNFR1-PKA interaction was not observed in the absence of TNF-α, we also questioned whether this was because of conformational change in TNFR1 induced by ligand treatment, or TNF-α-mediated activation of PKA. Forskolin treatment alone was not sufficient to promote TNFR1-PKA interaction, but it increased binding strength in the presence of TNF-α (Fig. 1D, E), suggesting a TNF-α-mediated conformational change in TNFR1 permitting PKA binding. After establishing binding, we next tested whether PKA directly phosphorylates TNFR1. To show this, we immunoprecipitated TNFR1 from serum-starved 293T cells, transfected with wild-type TNFR1 expression vector, and performed an in vitro kinase reaction using PKAcα and immunoblotted with anti-PKA substrate antibody. As given in Fig. 1F, there is a substantial amount of PKA-site phosphorylation of TNFR1 and addition of constitutively active PKAcα increased this by 2-fold. Phosphorylation of the RxxT motifs recognized by PKA substrate antibody was inhibited in the presence of PKI, validating phosphorylation of these residues by PKA.
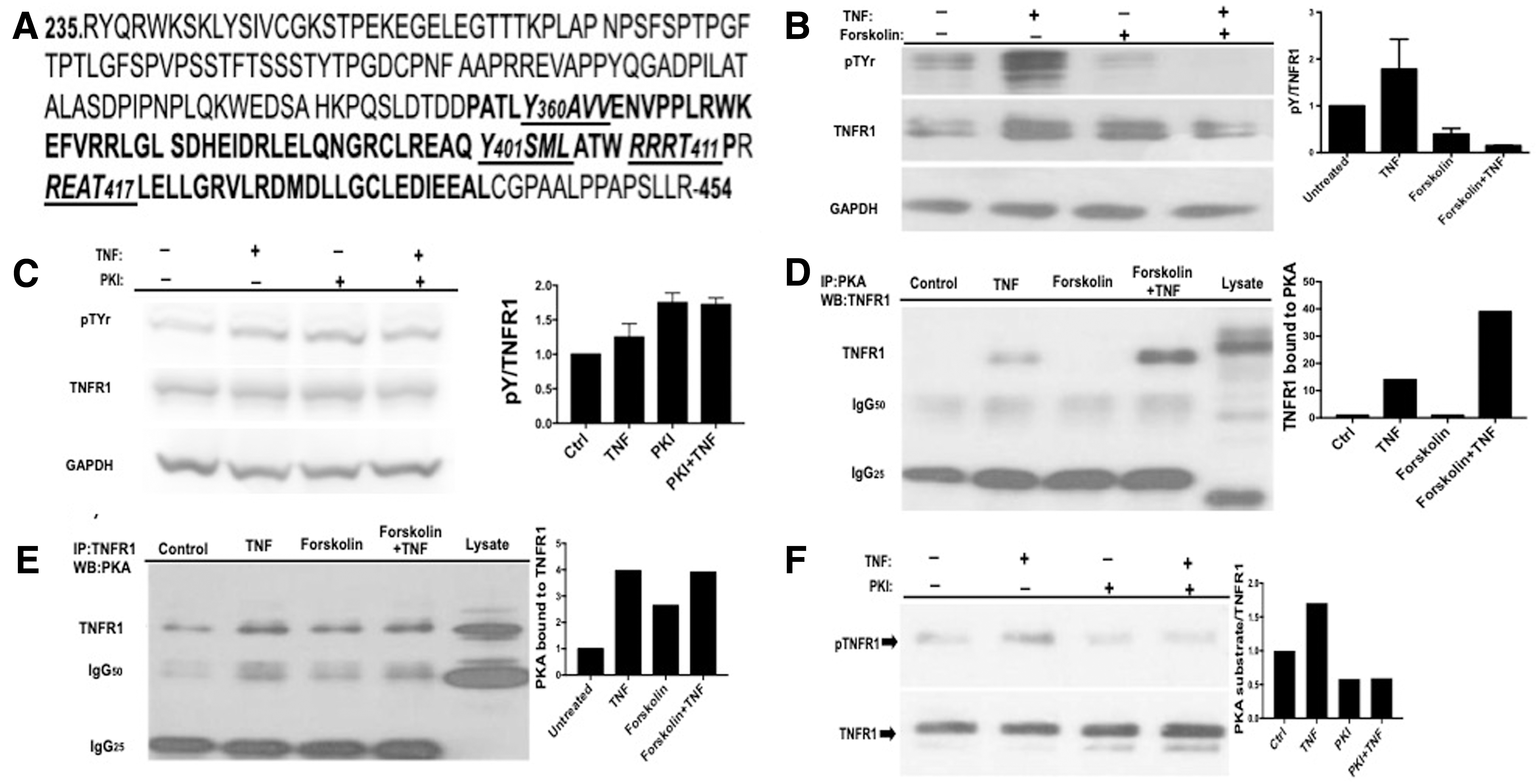
PKA activation inhibits TNF-α-induced Tyrosine phosphorylation.
TNF-α-induced phosphorylation of ERK1/2 is inhibited by PKA-mediated phosphorylation of TNFR1
To investigate the role of PKA-induced phosphorylations on per se “noncanonical TNF-α signaling,” we decided to look at the TNF-α-mediated phosphorylations of ERK, AKT, and STAT3 whose phosphorylations simply cannot be explained by the formation of complex-I. We started off with ERK phosphorylation and determined the time point of TNF-α-induced ERK phosphorylation in 293T cells. As given in Fig. 2A, TNF-α-induced ERK phosphorylation is very transient, reached a maximum level at 15th minute, and stay above basal level for at least 90 minutes. Because basal TNFR1 expression level is low in 293T cells and transfection of these cells by TNFR1 plasmids can increase TNFR1 expression by 25–30-fold, we decided to test the impact of PKA phosphorylation on TNFR1 signaling by transfecting TNFR1 mutants mimicking (Fig. 2C) or inhibiting (Fig. 2B) PKA phosphorylation. As maximum ERK activation was observed at 15th minute of TNF-α treatment (Fig. 2A), we tested the impact of our mutants on ERK phosphorylation after 15 min of TNF-α stimulation. As given in Fig. 2B, TNF-α induced 8-fold increase in ERK phosphorylation and this level was further increased to 17 and 23-fold in 293T cells transfected with T417A and T411/417A mutants, respectively. Moreover, alanine mutants showed positive additive effect on basal level of ERK phosphorylation. Although T411A mutant did not positively affect TNF-α-induced ERK phosphorylation, T411D mutant reduced TNF-α-induced ERK phosphorylation >50%; however, T417D and double mutant did not show significant effect (Fig. 2C). Based on these results, it is reasonable to say that phosphorylations of T411 and T417 of TNFR1 negatively affect TNF-α-induced ERK phosphorylation. To better understand the reason behind these observations, and because ERK phosphorylation requires Grb2 binding to TNFR1 (Hapil and others, submitted), we looked at the binding of Grb2 to wild type and mutants TNFR1. As shown at Fig. 2D and E, Grb2 constitutively binds to TNFR1 and addition of TNF-α slightly increases this. When we determined the binding of Grb2 to mutants, we found that Alanine mutants positively and phosphomimetic mutants negatively affected Grb2 binding. These results simply indicate that Grb2 binding to C-terminal Grb2-binding site (P448PAP451) is sensitive to T411 and T417 phosphorylations. Given the close proximity of Grb2-binding site to PKA phosphorylation sites, it is highly likely that T411 and T417 phosphorylations either perturb the structure of Grb2-binding site or create sterical hindrance due to electrostatics repulsion.

PKA phosphorylation-site mutants differentially affect TNF-α-induced ERK phosphorylation.
TNF-α-induced phosphorylation of Akt is inhibited by PKA-mediated phosphorylation of TNFR1
Although TNFR1 does not bear a YxxM motif (a binding site for the PI3K p85α sub unit), it has been known for a long time that TNFR1 binds to PI3Kp85α and transiently induces AKT phosphorylation (Ozes and others 1999, 2001; Pincheira and others 2008). Because we have shown previously that ERK phosphorylations are affected from T411/T417 phosphorylations, we wanted to determine whether TNF-α-induced AKT phosphorylation is also affected from PKA-induced phosphorylations of T411 and T417. Before testing the effects of our mutants, we determined time-dependent phosphorylation of AKT after TNF-α stimulation. In 293T cells, transfected with wild-type TNFR1 expression vector, TNF-α-induced maximum 2.2-fold AKT phosphorylation at 45th minute (Fig. 3A). Therefore, serum-starved 293T cells that ectopically expressed wild-type and mutant forms of TNFR1 were treated with TNF-α for 45 min. As given at Fig. 3B, T411A and T417A mutants positively stimulated both basal and TNF-α-induced AKT phosphorylation. However, all phosphomimetic mutants suppressed TNF-α-induced Akt phosphorylation (Fig. 3C), indicating that phosphorylations of T411 and T417 interferes with PI3K activation.
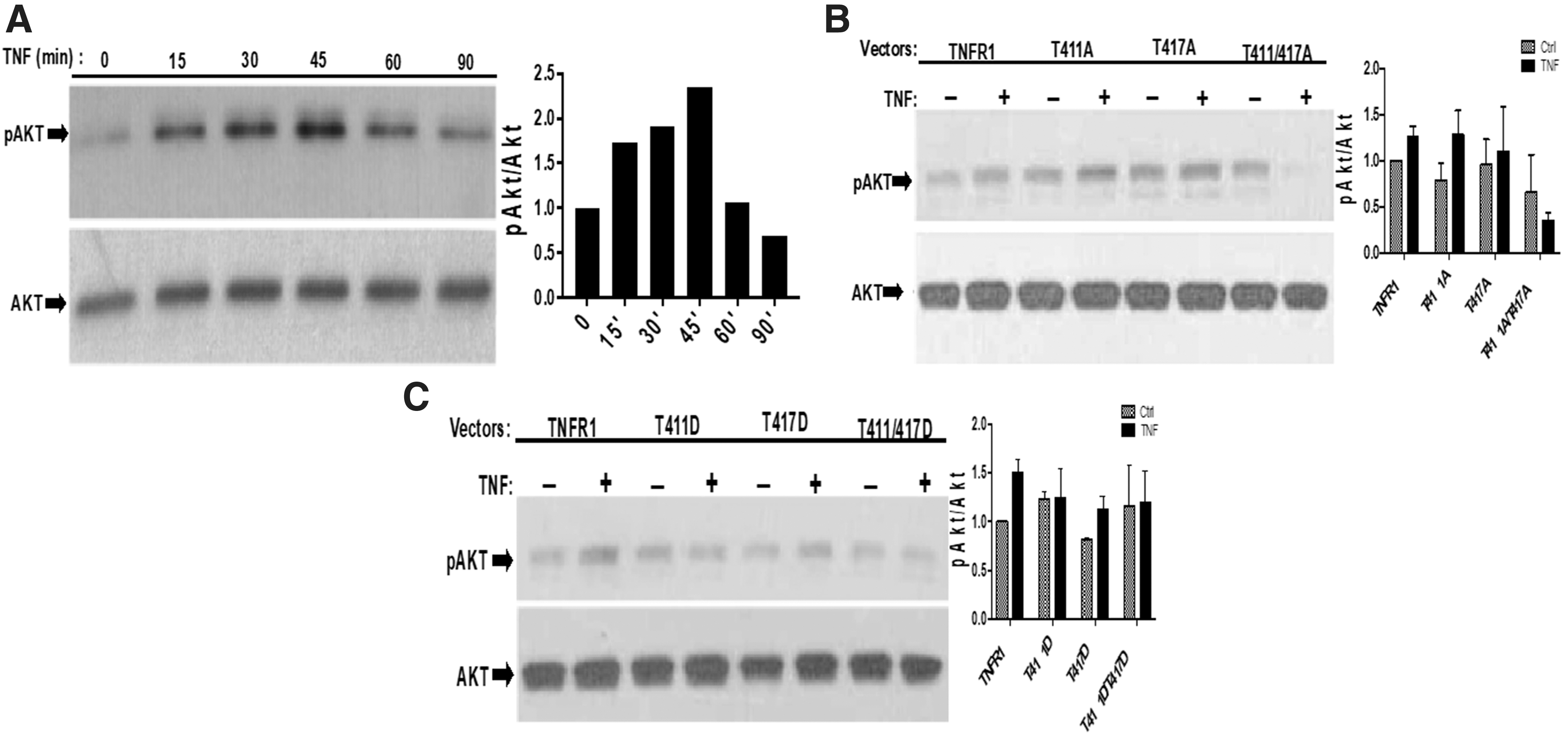
PKA phosphorylation-site mutants differentially affect TNF-α-induced Akt phosphorylation.
TNF-α-induced phosphorylation of STAT3 is inhibited by PKA-mediated phosphorylation of TNFR1
As mentioned previously, we believe that TNF-α-induced phosphorylations of ERK, AKT, and STAT3 cannot be explained by “canonical TNF signaling.” In line with this, to gain further insight about “noncanonical TNF-α signaling” we also determined the effect of PKA-site mutants on TNF-α-mediated tyrosine phosphorylation of STAT3. Once again, before testing this idea, we determined time-dependent tyrosine phosphorylation of STAT3 after TNF-α stimulation in 293T cells. As given at Fig. 4A, tyrosine phosphorylation of STAT3 is almost undetectable in untreated cells, and stimulation with TNF-α-induced maximum 17-fold STAT3 phosphorylation at 30th minute; therefore, we tested the effect of our mutants on STAT3 phosphorylation 30 minutes after TNF-α stimulation. Ectopic expression of T411D, T417D, and double mutant negatively affected tyrosine phosphorylation of STAT3, and double mutant almost completely eliminated basal level of STAT3 phosphorylation (Fig. 4B). However, contrary to our expectation, T411A, T417A, and double alanine mutants produced very similar results (Fig. 4C). Based on these, it is possible to argue that alanine mutants are artificial and may perturb the structure of C-terminal of TNFR1 and prevent STAT3 binding. However, it is commonly accepted that phosphomimetic mutants represent actual phosphorylation very well. Therefore, our results indicate that binding of STAT3 to TNFR1 is definitely affected from PKA-mediated phosphorylations of T411 and T417. In fact, we think that STAT3 may recognize C-terminal SH3-recognition site, P448PAP451 by its SH3 domain, just like Grb2.
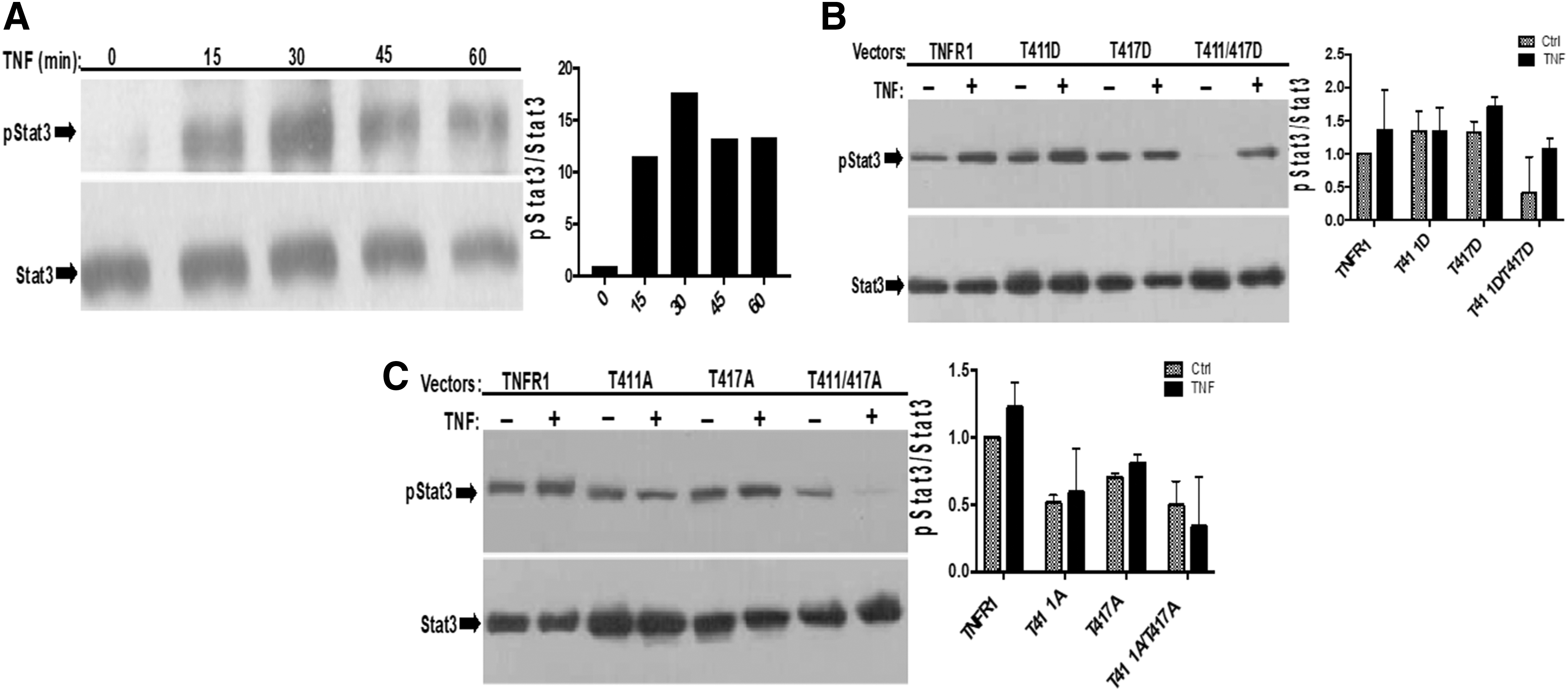
PKA phosphorylation-site mutants differentially affect TNF-α-induced STAT3 phosphorylation.
TNF-α-induced phosphorylation of JNK1/2 is inhibited by PKA-mediated phosphorylation of TNFR1
According to the “canonical TNFR1 signaling,” formation of complex-I and subsequent activation of TAK1 results in TAK1-mediated phosphorylation/activation of MAPKKs (mitogen-activated kinase kinase), MKK4/7. Activated MKK4/7 phosphorylates and activates all forms of JNKs. To gain further insight as to whether our PKA-phosphorylation site mutants would affect TNFR1-induced activations of JNK1/2, we determined the phosphorylation levels of these kinases. As given in Fig. 5A, there is very low level of JNK phosphorylation in unstimulated 293T cells and TNF-α induced maximum 13-fold JNK1/2 phosphorylation at 15th minute. Therefore, we tested the effect of our mutants on JNK1/2 phosphorylation 15 min after TNF-α stimulation. Ectopic expression of wild-type TNFR1 induced basal level of phosphorylation of JNKs and TNF-α further increased this. Moreover, expressions of single alanine mutants significantly increased basal JNK1/2 phosphorylations, however, did not further increase TNF-α-induced JNK1/2 phosphorylations. Contrary to alanine mutants, phosphomimetic mutants did not significantly affect basal level of TNF-α-induced JNK1/2 phosphorylations, although all double mutants severely diminished basal and TNF-α-induced JNK1/2 phosphorylations (Fig. 5B and C).

PKA phosphorylation-site mutants differentially affect TNF-α-induced JNK1/2 phosphorylation.
TNF-α-induced phosphorylation of p38α is inhibited by PKA-mediated phosphorylation of TNFR1
Because TNF-α-induced phosphorylations of p38 members are also mediated by TAK1-MKK3/6-p38 cascade, we also determined the impact of our mutants on TNF-α-induced phosphorylation/activation of p38α. Similar to JNK phosphorylation pattern, TNF-α induced maximum 5-fold p38α phosphorylation at 15th minute (Fig. 6A); therefore, we tested the effect of our mutants on p38α phosphorylation 15 min after TNF-α stimulation. As given in Fig. 6B and C, all mutants of TNFR1 significantly diminished TNF-α-induced phosphorylation of p38α, indicating that modification of PKA phosphorylation sites are interfering with canonical TNFR1 signaling possibly by influencing formation of complex-I.

PKA phosphorylation-site mutants differentially affect TNF-α-induced p38a phosphorylation.
TNF-α-induced activation of Caspase 8 is inhibited while necrosis is induced by PKA-mediated phosphorylation of TNFR1
As it is largely accepted, TNF-α-induced TNFR1 signaling is a very transient event and terminated owing to deubiquitination of TRADD, RIP1, NEMO, and TRAF2 by deubiquitinases A20 and CYLD (Opipari and others 1990; Trompouki and others 2003; Malynn and Ma 2009; Verhelst and others 2014; Legarda and others 2016). Deubiquitination of RIP1, NEMO, and TRAF results in dismantlement of complex-I; as a result of this, TRADD and RIP1 translocate to cytoplasm and participate in the formation of complex-IIa and complex-IIb, respectively. Formation of these complexes results in activation of Caspase 8 and MLKL and induction of apoptosis and necrosis, respectively (Varfolomeev and others 2008; Kalliolias and Ivashkiv 2016; Tseng and others 2018). According to these models, it was suggested that TNFR1 is not physically involved in induction of apoptosis or necrosis. Having all these in mind, we wanted to test the impact of our PKA-phosphorylation site mutants on basal and TNF-α-induced apoptosis and necrosis. As given in Fig. 7A ectopic expression of all mutants, except T411A, diminished basal level and inhibited TNF-α-induced induction of Caspase 8 activation. T411A mutant also decreased TNF-induced necrosis, which can also be because of increased late-phase of apoptosis (Fig. 7B). On the contrary, T417D and T411/417D expressing cells were more prone to TNF-induced apoptosis compared with cells expressing wild-type TNFR1 (Fig. 7B). These results clearly indicate that modification of T411 and T417 interfere with TNFR1 signaling possibly by modulating the formation of complex-I.

PKA phosphorylation-site mutants differentially affect TNF-α-induced Caspase 8 activation and induction of necrosis.
Discussion
In this study, we show for the first time that TNFR1 possesses 2 putative PKA phosphorylation motifs (RXXR, RRXR) and PKA negatively regulates TNFR1 signaling by phosphorylating these sites. The majority of the biological effects of TNF-α are mediated by signaling pathways initiated from TNFR1, and the presence of DD is essential for this. In the past 30 years, ground-breaking studies identified the proteins involved in initiation and transduction of TNF-α signaling. All these studies elegantly elucidated sequential bindings of TRADD-RIP1-TRAF2-cIAP1/2 and LUBAC to TNFR1 and subsequent ubiquitination of TRADD, RIP1, NEMO, and TRAF2 in this complex. These ubiquitinations create docking sites for TAB2/3-TAK1 and NEMO-IKKα-IKKβ complexes, resulting in formation of complex-I. The formation of complex-I induces autophosphorylation and activation of TAK1, which in turn phosphorylates and activates IKKs as well as MKK3/6 and MKK4/7. Activated IKKs induce NF-kB activation by phosphorylating IkBs, whereas MKK3/6 and MKK4/7 phosphorylate and activate p38s and JNKs, respectively (Hsu and others 1995, 1996a, 1996b; Rothe and others 1995; Shu and others 1996; Wang and others 2001; Park and others 2004; Ea and others 2006; Zheng and others 2006; Jackson-Bernitsas and others 2007; Bertrand and others 2008; Varfolomeev and others 2008; Haas and others 2009; Tokunaga and others 2009; Gerlach and others 2011). All these signaling events are negatively regulated or terminated by deubiquitination of TRADD, RIP1, NEMO, and TRAF2 by deubiquitinases A20 and CYLD (Opipari and others 1990; Trompouki and others 2003; Malynn and Ma 2009; Verhelst and others 2014; Legarda and others 2016). Although the formation of complex-I can explain TNF-α-induced activations of NF-kB, p38s and JNKs, it cannot explain as to how TNF-α induces phosphorylations/activations of AKT, ERK, and STAT3 as shown by us and others (Guo and others 1998; Ozes and others 1999, 2001; Miscia and others 2002; Pincheira and others 2008; Ma and others 2017). In line with this, we have recently shown, for the first time, that TNFR1 is phosphorylated at Y360 and Y401 by JAK2 and c-SRC upon TNF-α stimulation, and we believe that these phosphorylations underline the mechanism of TNF-α-induced phosphorylations/activations of AKT, ERK, and STAT3 (Hapil and others, submitted).
Apart from the modulation of TNF-α signaling by A20 and CYLD, almost nothing is known about negative regulation of TNFR1 signaling, at least at the molecular level. However, it has been known for a long time that cyclic-AMP-inducing factors and activation of cAMP-activated kinase (PKA) or inhibition of phosphodiesterases increase cellular cyclic AMP level, interfere with production of TNF-α or negatively affect TNF-α-induced induction or activations of several end points including inflammation, NF-kB activation, ICAM induction, and so on (Strieter and others 1988; Endres and others 1991; Matuschak and others 1994; Neumann and others 1995; Ollivier and others 1996; Takahashi and others 2002; Coimbra and others 2005, 2006; Stafford and Marnett 2008). To elucidate the molecular mechanism behind all these observation, we searched amino acid sequence of intracytoplasmic portion of TNFR1 and found 2 putative PKA phosphorylation sites RRRT411 and REAT417 (Fig. 1A). Before starting molecular studies, we first showed that TNF-α-induced tyrosine phosphorylation of TNFR1 is severely inhibited by pretreatment of cells with PKA-activator Forskolin (Fig. 1B). To understand the mechanism behind this observation, we performed immunoprecipitation and in vitro kinase assays and showed that PKAcα subunit strongly binds to TNFR1 only after TNF-α stimulation and directly phosphorylates it (Fig. 1C and D). Given the fact that TNF-α by itself induces activation of PKA (El-Ani and others 2014), our results strongly suggest that TNF-α-mediated activation of PKA is serving as a negative feedback loop mechanism to prevent continuous TNF-α signaling, which is pathogenic. After establishing direct functional relationship between TNFR1 and PKA, we wanted to have genetic evidences to support these findings. For this, we have generated alanine and aspartic acid mutants of human TNFR1 expression vector at T411 and T417, ectopically expressed these mutants along with wild-type TNFR1, and determined the level of TNF-α-induced phosphorylations of ERK, AKT, STAT3, p38α, and JNK1/2. In addition to these, we also determined the levels of Caspase 8 activation and induction of necrosis.
As mentioned previously, we strongly believe that TNF-α-induced phosphorylations of ERK, AKT, and STAT3 are dependent on tyrosine phosphorylation of TNFR1 at Y360 and Y401 (Hapil and others, submitted), and most likely independent of complex-I formation; therefore, we call this route of TNF-α signaling as “noncanonical TNF-α signaling pathway.” Because PKA activation severely inhibits tyrosine phosphorylation of TNFR1, we thought that phosphorylation of TNFR1 at T411 and T417 could be mimicked by our aspartic acid mutants; therefore, we anticipated that ectopic expression of these phosphorylation mimicking mutants would negatively affect TNF-α-induced phosphorylations of ERK, AKT, and STAT3, whereas alanine mutants would augment phosphorylations of these. Indeed, as given in Figs. 2–4, there is a strong trend toward augmentation of TNF-α-induced phosphorylation of ERK, AKT, and STAT3 in 293T cells ectopically expressing T411A, T417A, and T411/417A mutants, whereas these phosphorylations were suppressed in 293T cells ectopically expressing T411D, T417D, and T411/417D mutants.
Overall, these results can be explained in a way that PKA-mediated phosphorylations of TNFR1 at T411 and T417 can change the folding of TNFR1 in such a way that (1) distorted structure of TNFR1 becomes inaccessible for bindings of JAK2 and c-SRC, (2) distorted structure of TNFR1 can accommodate stronger bindings of tyrosine phosphatases, such as SHP2 to TNFR1, resulting in dephosphorylation of p-Y360 and p-Y401, (3) binding of GRB2 to PPAP (SH3 recognition sequence) (Hildt and Oess 1999) of TNFR1 by SH3 domain of GRB2 is prevented because of electrostatic repulsion given the very close proximity of PPAP to T411 and T417. In fact, our findings (Fig. 2D and E) strongly support this hypothesis. The binding of GRB2 to PPAP sequence can support Grb2-SOS-Ras-Raf-Erk and Grb2-SOS-Ras-PI3kp100-Akt (Rodriguez-Viciana and others 1994) signal transduction pathways. Given the fact that TNF-α-induced Stat3 tyrosine phosphorylation is mediated by c-SRC (Pincheira and others 2008), and PPAP sequence can be used for docking of c-Src and Stat3, PKA-mediated phosphorylation of T411 and T417 seem to be affecting these dockings and subsequent phosphorylations. Overall, PPAP sequence seems to control TNF-α-induced phosphorylations of ERK, AKT, and STAT3 simply by serving as docking sites for Grb2, c-SRC, and Stat3.
Although we have anticipated to see what we observed in terms of TNF-α-induced activations of Erk, Akt, and Stat3 are concerned, we did not know how modifications of T411 and T417 would affect downstream elements of complex-I. To our surprise, as shown in Figs. 5–7A, ectopic expressions of all PKA-site mutants significantly interfered with TNF-α-induced activations of JNK1/2, p38α, and Caspase 8, respectively. Contrary to these, the same mutants augmented induction of necrosis in a positive-additive way (Fig. 7B). Because activations of JNK1/2, p38α, and Caspase 8 requires formation of complex-I, whereas induction of necrosis requires disintegration/dissolution of complex, our results suggest that PKA-mediated phosphorylation of TNFR1 can cause a conformational change in the structure of TNFR1 in such a way that complex-I can no longer be formed and dissociated RIP1 moves to cytoplasm and participates in the formation of necrosis-inducing complex.
Altogether, our findings strongly suggest that TNF-α-activated PKA phosphorylates T411 and T417, this modifies the structure of TNFR1 in a way that canonical and noncanonical TNF-α signaling components can no longer bind to TNFR1, and as a result, all TNFR1-based signal transductions are terminated. This negative feedback mechanism complements the activity of A20 and CYLD, and explains long known anti-TNF-α and anti-inflammatory activity of PKA.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by TUBITAK [Grant No. 113s335] and Akdeniz University Scientific Research Project Coordination Unit [Grant No. TDK-2016-1272].