
Abstract
The balance between inflammatory and anti-inflammatory immune responses is maintained through immunoregulatory cell populations and immunosuppressive cytokines. Interleukin-35 (IL-35), an inhibitory cytokine that belongs to the IL-12 family, is capable of potently suppressing T cell proliferation and inducing IL-35-producing induced regulatory T cells (iTr35) to limit inflammatory responses. Over the past decade, a growing number of studies have indicated that IL-35 plays an important role in controlling immune-related disorders, including autoimmune diseases, infectious diseases, and cancer. In this review, we summarize the current knowledge about the biology of IL-35 and its contribution in different diseases, and we discuss the potential of and barriers to harnessing IL-35 as a clinical biomarker or immunotherapy.
Introduction
Interleukin-35 (IL-35) is a recently identified cytokine in the IL-12 family, which plays a key role in the suppressive function of regulatory T cells (Tregs) (Collison and others 2007, 2010). IL-12 family cytokines are highly promiscuous and produced as heterodimers composed of an alpha-chain [p19 (encoded by Il23a), p28 (encoded by Il27a), or p35 (encoded by Il12a)] and a beta-chain [p40 (encoded by Il12b) or Epstein-Barr virus-induced 3 (Ebi3, encoded by Ebi3)] (Vignali and Kuchroo 2012). The IL-12 family is currently composed of 4 main heterodimeric cytokines, including IL-12 (p35 and p40) (Kobayashi and others 1989), IL-23 (p19 and p40) (Oppmann and others 2000), IL-27 (p28 and Ebi3) (Pflanz and others 2002), and IL-35 (p35 and Ebi3) (Fig. 1) (Collison and others 2007). A fifth was recently proposed, IL-39 (p19 and Ebi3) (Wang and others 2016b).
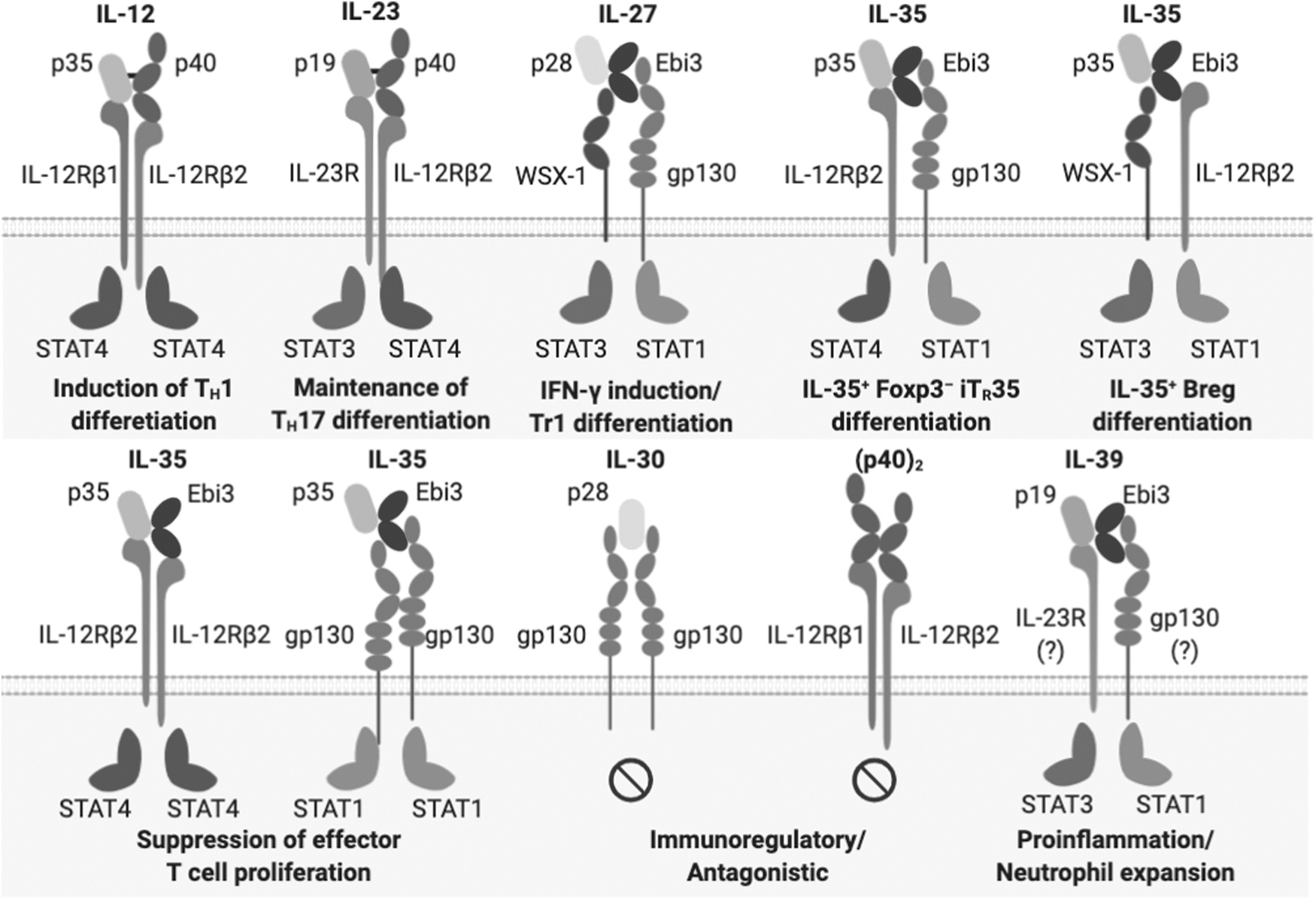
IL-35 is a member of the promiscuous IL-12 family of cytokines and signaling receptors. Family members are categorized into 2 distinct subunit chain types based on their structural homology: alpha-chains (p19, p28, and p35) and beta-chains (p40 and Ebi3). Alpha-chains also belong to the IL-6 superfamily due to their conserved 4 alpha helix-bundle structure. Beta-chains show structural homology to IL-6Rα and are also members of the class I cytokine receptor family (homology based on tandem fibronectin type III domains and conserved WSXWS motif near cytokine binding site). Their physiological roles range from potent proinflammatory immune responses (IL-12/IL-23/IL-27/IL-39) to immune regulation [IL-27/IL-35/IL-30/(p40)2]. Ebi3, Epstein-Barr virus induced 3; IL, interleukin.
Although the other cytokines in the IL-12 family are mainly produced by activated antigen-presenting cells (APCs) such as monocytes, dendritic cells (DCs), and macrophages, IL-35 was initially found to be produced by activated Tregs. Subsequent studies reported the expression of IL-35 in several other immunoregulatory cells (Fig. 2), such as regulatory B cells (Bregs) (Shen and others 2014; Wang and others 2014b), tolerogenic DCs (tolDCs) (Dixon and others 2015; Haller and others 2017; Koga and others 2021), pro-metastatic tumor-associated macrophages (Lee and others 2018), and CD8+ Tregs (Olson and others 2012). Numerous studies have demonstrated the significance of IL-35, with its potent suppressive function, in a variety of diseases including cancer, autoimmunity, and infections (Table 1).
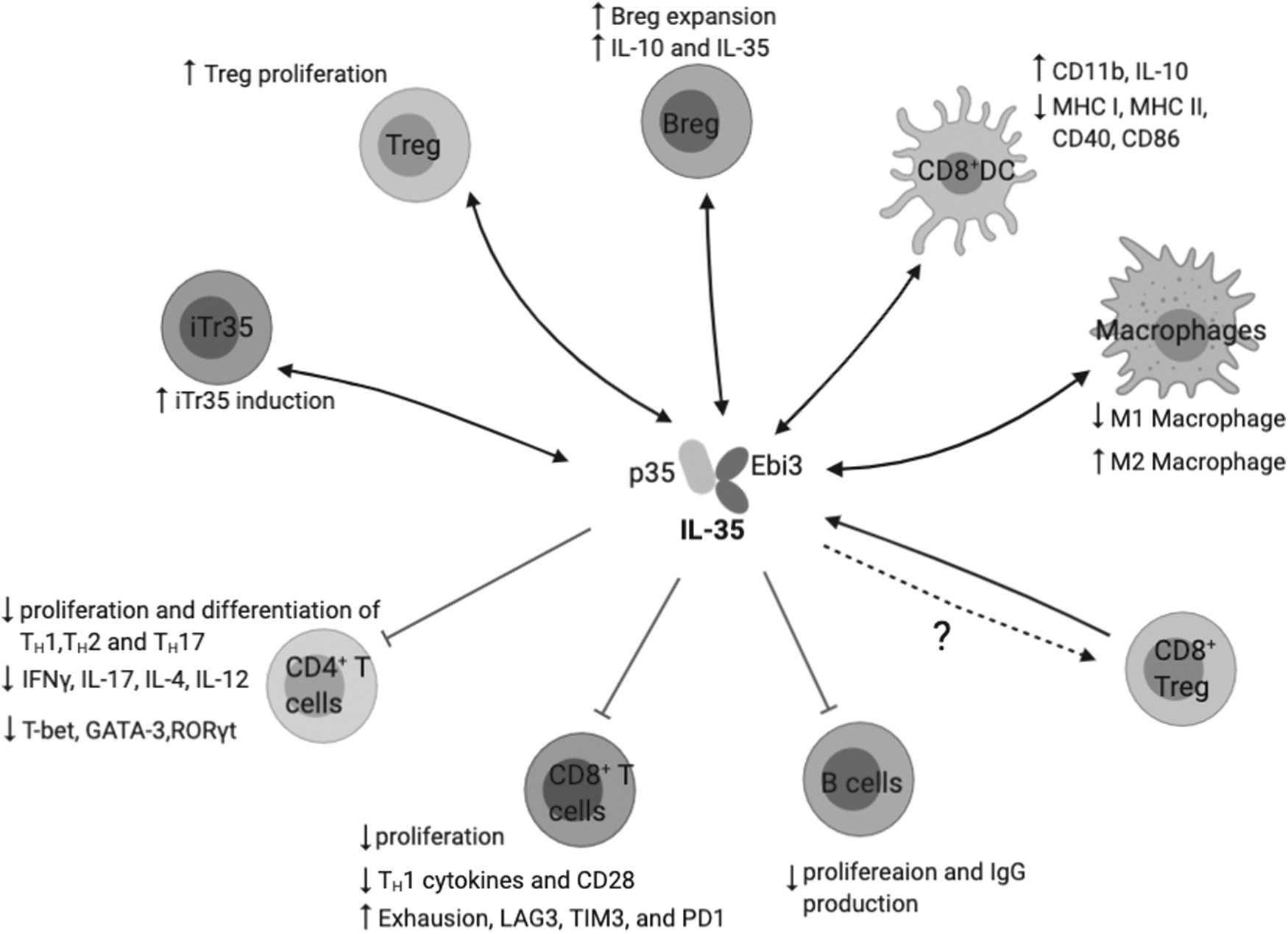
Summary of IL-35 responders and producers. IL-35 is produced by a wide range of immune cells, including Tregs, iTr35, Bregs, tolerogenic DCs, and macrophages, and they primarily mediate suppression on effector T cells and B cells. Bregs, regulatory B cells; DCs, dendritic cells; iTr35, IL-35-producing induced regulatory T cells; Tregs, regulatory T cells.
Summary of Interleukin-35 Involvement and Impact in Various Diseases
Bregs, regulatory B cells; CIA, collagen-induced arthritis; DCs, dendritic cells; EAE, experimental autoimmune encephalomyelitis; Ebi3, Epstein-Barr virus induced 3; HBV, hepatitis B virus; HCV, hepatitis C virus; IBD, inflammatory bowel disease; IFN, interferon; IL, interleukin; iTr35, IL-35-producing induced regulatory T cells; MS, multiple sclerosis; NOD, non-obese diabetic; pSS, primary Sjögren syndrome; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SNP, single nucleotide polymorphism; T1D, type 1 diabetes; Treg, regulatory T cell.
In this review, we will discuss what is currently known about IL-35 expression and its signaling pathway as well as the physiological importance of IL-35 in autoimmunity, cancer, and infectious diseases. Finally, we will also discuss the obstacles and the potentials in targeting IL-35 as a novel therapeutic approach and other potential IL-12 family members.
The Composition of IL-35
Although the interaction between Ebi3 and p35 as a secreted heterodimeric complex was first reported in 1997 in various cell lines and human placenta, its physiological role was not determined (Devergne and others 1997). In 2007, it was shown that an Ebi3–p35 heterodimer, which was named IL-35, is produced by activated Tregs and suppresses T cell-driven activity, such as the homeostatic expansion of T cells in lymphopenic mice and inflammatory bowel disease (IBD) (Collison and others 2007). More recently, a subset of B cells that produce IL-35 was identified and shown to provide protection against autoimmune diseases and suppress B220hi B cell proliferation (Shen and others 2014; Wang and others 2014b). In addition, the expression of IL-35 in tolDCs (Dixon and others 2015), pro-metastatic tumor-associated macrophages (Lee and others 2018), and CD8+ Tregs (Olson and others 2012) has been suggested, implicating its importance in a wide range of suppressive cell populations, diseases, and inflammatory environments.
Ebi3 was first cloned as a gene induced by Epstein-Barr virus (EBV) infection in B cells and shown to encode a 34-kDa glycoprotein (Devergne and others 1996, 1997). It was initially characterized as a member of the IL-6 receptor superfamily with the closest homology to ciliary neurotrophic factor receptor (30% identity) and IL-12p40 (27% identity) (Devergne and others 1996). Indeed, Ebi3 is structurally homologous to the receptors of IL-6 cytokines and belongs to the type I cytokine receptor superfamily as it contains fibronectin type III domains as well as LDSWS motif, which is closely related to a highly conserved WSXWS motif that contributes to ligand binding of type I cytokine receptors (Devergne and others 1996; Jones and Vignali 2011; Dagil and others 2012; Hasegawa and others 2016). However, Ebi3 is secreted without post-translational proteolytic modification as it lacks transmembrane and signaling domains (Hasegawa and others 2016).
The expression of Ebi3 is mainly enriched in myeloid cells, especially upon activation of the transcription factors NFκB and PU.1, and stimulation of toll-like receptor (TLR) by endotoxins, such as lipopolysaccharides (Pflanz and others 2002; Wirtz and others 2005; Cassatella and others 2020). In addition, various CD4+ T cell subsets and other peripheral blood cells upregulate Ebi3 expression upon activation, such as plate-bound anti-CD3/CD28 or pokeweed mitogen (Devergne and others 1996; Ma and others 2019).
The other component of IL-35, the IL-12p35 chain, was first reported as a 35 kDa component of natural killer cell stimulatory factor, which was later named IL-12, produced by EBV-transformed human B cells (Kobayashi and others 1989). Subsequent studies have established that IL-12 plays a crucial role in triggering type 1 inflammation and facilitates the formation of a feedback loop between APCs and naive T cells for appropriate Th1 differentiation (Shu and others 1995; Cella and others 1996; Ma and others 1996). Unlike Ebi3, Il12a gene expression requires a combination of NFκB and IRF-1 activated by simultaneous stimulation of APCs with endotoxin and interferon-γ (IFN-γ), respectively (Liu and others 2003).
Moreover, the expression of p35 is under the control of both intrinsic and extrinsic post-translational regulations. Unlike its alpha-chain relative, p28, which can be secreted as a monomeric form independent of its binding partner in mice (IL-30) (Crabe and others 2009; Stumhofer and others 2010; Garbers and others 2013), p35 accumulates in the endoplasmic reticulum (ER) in the absence of its partner beta-chains (Reitberger and others 2017). Murine p28 contains a unique loop domain containing 13 consecutive glutamic acid repeat (poly-Glu loop) that is not conserved in any of the other alpha-helix bundle cytokine chains (Reitberger and others 2017). The hydrophobic interaction between the poly-Glu loop and AB-loop results in an intramolecular disulfide bond. This self-sustained protein folding facilitates a unique partner-less secretion of p28 monomers, which is referred to as IL-30 (Stumhofer and others 2010; Dibra and others 2012). In contrast, p35 lacks this hydrophobic loop–loop interaction and forms erroneous disulfide bonds that cause misfolding in the ER. However, the presence of partner beta-chains facilitated the formation of native disulfide bonds in p35 and subsequent secretion as a heterodimer complex (Reitberger and others 2017). Thus, the self-limiting misfolding-prone protein structure of p35 tightly regulates its bioavailability and bioactivity.
IL-35 Expression Pattern
Although p35 expression is largely restricted to APCs, CD4+CD25+ Tregs were shown to co-express Ebi3 and p35 in a Foxp3-dependent manner (Collison and others 2007; Gavin and others 2007). Further investigation revealed that Tregs do not express other remaining alpha-chains of IL-12 family (p19 and p28), indicating that IL-35 is the only member of IL-12 family cytokines produced by Tregs (Collison and others 2007). Although it is unclear whether NFκB, PU.1, and IRF-1 contribute to IL-35 expression in Tregs, TCR stimulation was shown to be an essential driver as activated Tregs preferentially expressed both Ebi3 and Il12a (Collison and others 2007, 2009).
Moreover, the presence of effector T cells upon in vitro activation of Tregs highly upregulated Ebi3 and Il12a expression, whereas the absence of effector T cells resulted in the downregulation of the genes (Collison and others 2009). This observation suggests that Tregs require additional cell-contact dependent stimulation to direct the TCR downstream signaling to promote IL-35 expression, which was subsequently found to be driven by a Sema4a–Nrp1 axis, which is required and sufficient for contact-independent suppressive activity of activated Tregs (Delgoffe and others 2013; Overacre-Delgoffe and others 2017). However, it is noteworthy that only a proportion of Foxp3+ Tregs produce IL-35 (Wei and others 2017; Sawant and others 2019), despite their high expression level of Nrp1; thus, the exact downstream mechanism(s) by which Tregs express Ebi3 and p35 has not been fully elucidated. As IL-35 expression has been reported in different diseases affecting various tissues and types of inflammation, it is possible that other environmental factors, such as cytokines, chemokines, growth factors, and metabolites, individually or collectively promote IL-35 expression in a context-dependent manner.
IL-35 Heterodimer Formation
Despite the structural homology between Ebi3 and p40, Ebi3-derived cytokines (IL-27 and IL-35) and p40-derived cytokines (IL-12 and IL-23) are structurally and functionally distinct. Within the IL-12 or IL-23 complex, the p40 subunit forms a disulfide bond with alpha-chain partners via a cysteine residue in the AB-loop domain, contributing to the extracellular stability of secreted heterodimers (Lupardus and Garcia 2008; Reitberger and others 2017). On the other hand, Ebi3 does not form a disulfide bond with either of its binding partners (Aparicio-Siegmund and others 2014). Thus, Ebi3-derived cytokines (IL-27 and IL-35) are less stable with reduced secretion and extracellular stability compared with IL-12 and IL-23 (Jones and Vignali 2011; Vignali and Kuchroo 2012). However, it remains elusive whether the increased stability of IL-12 would result in a preferential production of IL-12 over IL-35 when p35, p40, and Ebi3 are co-expressed (Jones and Vignali 2011).
Due to this instability, the crystal structure of IL-27 and IL-35 is still unknown, which makes it difficult to decipher the pairing of subunits (p28, p35, and Ebi3). Meanwhile, a mutagenesis study showed that a large array of p35 or Ebi3 mutants involved dimerization to form IL-12 and/or IL-27 fails to prevent the generation of an IL-35 heterodimer, suggesting that the interaction of Ebi3 and p35 is unique and different from other IL-12 members (Jones and others 2012). Although the pairing of the IL-35 warrants further investigation, a predicted model with 3 interaction sites has been proposed to explain the pairing in the IL-12 family based on IL-6 and leukemia inhibitory factor (LIF) complex crystal structures (Jones and Vignali 2011). Although site 1 regulates the interaction of heterodimer subunits, the α-helical subunit binds to the cytokine-binding homology region of the receptor chain through site 2 and interacts with the Ig domain of a second receptor chain via site 3 (Jones and Vignali 2011; Vignali and Kuchroo 2012).
IL-35 Receptors and Signaling Pathway
The IL-35 is highly promiscuous and unconventional in the number and types of receptors it utilizes and the signaling pathways it induces (Collison and others 2012; Wang and others 2014b). IL-35 shares its receptor chains with IL-12 and IL-27 in a manner analogous to how it shares cytokine subunit chains (Fig. 1) (Collison and others 2012; Vignali and Kuchroo 2012; Wang and others 2014b). The IL-35 receptors were first identified in T cells, which constitute an IL-12Rβ2/gp130 heterodimer or homodimers of either chain (Collison and others 2012). Although either IL-12Rβ2 or gp130 homodimers partially mediate IL-35-mediated immunosuppressive signaling, maximal function is elicited by the IL-12Rβ2/gp130 heterodimeric receptor (Collison and others 2012). Upon cytokine binding, the IL-35 receptors activate a signaling cascade via the Janus kinase–signal transducer and activator of transcription (JAK–STAT) pathway.
Although IL-12Rβ2/gp130 heterodimeric receptors signal through a novel STAT1/STAT4 transcription factor heterodimer complex in CD4+ T cells, the gp130 or IL-12Rβ2 homodimers elicits downstream signal transduction via activation of STAT1 or STAT4, respectively (Collison and others 2012; Murakami and others 2019). The heterodimerization of pSTAT1 and pSTAT4 promotes the expression of Ebi3 and Il12a by binding to their promoter regions, leading to the generation of Foxp3− IL-35-producing induced regulatory T cells (iTr35). This creates a positive feedback loop to further induce iTr35 cell differentiation, resulting in “infectious tolerance” (Collison and others 2010). Moreover, a recent study elucidated that IL-35-coated extracellular vesicles could cross-mark non-Treg cells as IL-35 “producers” and interact with bystander lymphocyte-expressing IL-35 receptors to promote “infectious tolerance,” highlighting an atypical mechanism through which IL-35 can mediate suppression and propagate tolerance (Sullivan and others 2020).
Interestingly, IL-35 can also utilize an alternative receptor composition to elicit signaling in B cells. For instance, blocking gp130 signaling had no effect on IL-35-mediated suppression of proliferation or IL-10 production in B cells, and further investigation demonstrated that B cells utilize IL-12Rβ2/WSX-1 (also known as IL-27Rα) heterodimers as the predominant IL-35 receptor (Wang and others 2014b). The interaction between IL-35 and the IL-12Rβ2/WSX-1 heterodimer results in the phosphorylation of STAT1 and STAT3 (Wang and others 2014b). IL-35-induced pSTAT1 in B cells directly binds to the Ebi3 and Il12a promoters, resulting in the differentiation of IL-35-producing Bregs. These observations suggest that Breg-derived IL-35 is also capable of triggering “infectious tolerance.” Further, IL-35 was still capable of mediating partial suppression of B cell proliferation upon in vitro knockdown of a single receptor chain (either WXS-1 or IL-12Rβ2). However, concomitant expression of both IL-12Rβ2 and WSX-1 was required to maximally suppress B cell proliferation and to induce the differentiation of IL-35+ Bregs (Wang and others 2014b), similarly to IL-35 signaling in CD4+ T cells (Collison and others 2012).
Although IL-12Rβ2 is expressed mainly by activated T cells, natural killer cells, B cells, and DCs (Presky and others 1996), gp130 is constitutively expressed in a wide range of cell types (Sawant and others 2015). Although IL-12Rβ2 is nearly undetectable on resting T cells, TCR-driven activation as well as co-stimulation with CD28 and cytokines, including IL-2, IL-12, IL-27, tumor-necrosis factor α (TNF-α), and IFN-γ, upregulate the expression of IL-12Rβ2 (Szabo and others 1997; Wu and others 2000; Collison and Vignali 2008). In addition, several reports have shown that WSX-1 is also broadly expressed by various cell types, including non-hematopoietic cells, such as epithelial cells (Diegelmann and others 2012; Cui and others 2017). Given the capability of IL-35 to signal through homodimers of IL-12Rβ2, gp130, it raises the possibility that IL-35 may also widely impact the non-hematopoietic compartment. Further, due to the chain-sharing feature of IL-12 family cytokines, IL-27 and IL-35 may compete for gp130 and WSX-1 receptor chains, whereas IL-12 and IL-35 compete for IL-12Rβ2.
Collectively, these results indicated that IL-35 can signal through at least 4 receptors (2 homodimers: IL-12Rβ2/IL-12Rβ2 and gp130/gp130, and 2 heterodimers: IL-12Rβ2/gp130 and IL-12Rβ2/WSX-1) and elicit at least 2 sets of transcription factor complex (STAT1/STAT4 and STAT1/STAT3).
IL-35 in Cancer
The tumor microenvironment (TME) (Mayer and others 2014), especially that of advanced cancers, is highly immunosuppressive and is equipped with various immunosuppressive mechanisms, one of which is the recruitment of immunoregulatory populations of immune cells that dampen anti-tumor immunity. Previous studies have shown that IL-35+ Tregs are highly enriched in the TME (Mayer and others 2014), and IL-35 blockade resulted in improved effector anti-tumor immune responses with subsequently reduced tumor burden and metastasis (Turnis and others 2016). More recently, it was revealed that Treg-derived IL-35 directly induces inhibitory receptor expression, while limiting the differentiation of a central memory CD8+ T cell population via a STAT1/4-BLIMP1 axis (Turnis and others 2016; Sawant and others 2019).
However, Tregs are not the only source of IL-35, and various cell types have been reported to produce IL-35 in the TME. For instance, it had been suggested that tumor cells in breast cancer and pancreatic ductal adenocarcinoma can produce IL-35 and that expression correlates with worse prognosis in patients with cancer (Hao and others 2018). A study utilizing a murine model of transplantable melanoma with transgenic IL-35 expression has demonstrated that increased cytokine production in the TME was associated with increased recruitment of myeloid-derived suppressor cells and angiogenesis, which collectively accelerated tumor growth (Wang and others 2013).
A recent study has suggested that metastatic TME-associated macrophages produce IL-35 in both humans (metastatic head and neck cancer) and mice (orthotopic mammary gland tumor model) (Lee and others 2018). Initial inflammation induced by tumor growth in situ upregulated IL-12Rβ2 expression in tumor cells to confer responsiveness to IL-35. Macrophage-derived IL-35 modulated the JAK2-STAT6-GATA3 axis to induce mesenchymal–epithelial transition in migrating tumor cells to establish metastasis (Lee and others 2018). Macrophage-derived IL-35 can suppress an anti-tumor response, whereas IL-35 from the TME can also promote the accumulation of myeloid-derived inhibitory cells, which can be ameliorated by IL-35 blockade (Zhu and others 2020; Wang and others 2021). Further, although B cell infiltration in the TME has been correlated with better patient prognosis in various cancer types, such as pancreatic cancer (Tewari and others 2013) and head and neck squamous cell carcinomas (Bruno and others 2017), a recent study demonstrated that IL-35+ Bregs promote cancer cell proliferation and accelerate the development of pancreatic ductal adenocarcinoma (Pylayeva-Gupta and others 2016).
These observations suggest that tumor-associated IL-35 enrichment is associated with pro-tumor immunoregulation and poses a barrier to effective cancer immunotherapy. Therefore, IL-35 is an appealing novel therapeutic target to treat cancer. However, the promiscuity of the IL-35 chain in sharing and utilization of multiple receptors complicates mechanistic dissection and therapeutic development.
As IL-35 receptor subunits are also shared among numerous cytokines within the IL-6/IL-12 cytokine family, it is imperative to understand the physiological role of each receptor chain. As discussed earlier, the IL-12Rβ2 chain is also utilized by IL-12 (Fig. 1), which is a crucial proinflammatory cytokine that has been associated with a beneficial clinical outcome in patients with colorectal cancer (Saito and others 2016). In addition, gp130 is ubiquitously expressed in both hematopoietic and non-hematopoietic compartments and used by many different cytokines other than IL-35, such as those in the IL-6 superfamily (Rose-John 2018; Murakami and others 2019), including IL-6 (Taga and others 1989) and LIF (Ip and others 1992).
Although IL-6 plays a detrimental role in facilitating autoimmune diseases such as arthritis and psoriasis (Ishihara and Hirano 2002), the increased expression of intratumoral IL-6 or circulating IL-6 has been correlated with poor survival in patients with various cancers (Fisher and others 2014; Kumari and others 2016; Vainer and others 2018). Further, LIF is a highly anti-inflammatory cytokine and is capable of skewing the CD4+ effector T cell differentiation toward Tregs (Janssens and others 2015). LIF is also highly upregulated in various types of cancer, and it can directly promote cancer cell survival and stemness via both STAT3-dependent and -independent pathways (McLean and others 2019).
Therefore, neutralization of IL-35 by monoclonal antibodies may prove to be an attractive immunotherapeutic approach. Indeed, we have previously demonstrated that it is possible to generate a monoclonal antibody against murine Ebi3 that selectively neutralizes the signal transduction of IL-35 while sparing that of IL-27 and that systemic neutralization of IL-35 recapitulated the reduced tumor burden observed in mice with Treg-restricted deletion of IL-35 without detectable immune-related adverse events (Turnis and others 2016). As there are no clinical trials currently examining the efficacy of IL-35 neutralization, the development of specific anti-human IL-35 therapeutics is warranted.
IL-35 in Autoimmunity and Inflammatory Diseases
Ectopic activation of effector immune responses and subsequent dysregulated self-tolerance result in many autoimmune and inflammatory diseases. Since its discovery, IL-35, as an important inhibitory cytokine, has been shown to mediate a protective role in a variety of autoimmune diseases. Interestingly, Ebi3- or p35-deficient mice do not spontaneously develop overt autoimmunity or inflammatory diseases (Mattner and others 1996; Nieuwenhuis and others 2002), despite the lack of IL-35. The lack of IL-35-mediated immunoregulation may be complicated by the utilization of Ebi3 and p35 by other known cytokines, such as IL-27 and IL-12, and perhaps as yet unknown cytokines. However, upon challenge, Ebi3- and p35-deficient mice exhibit exacerbated autoimmunity, such as experimental autoimmune encephalomyelitis (EAE) (Gran and others 2002; Liu and others 2012). These observations suggest that IL-35 plays a more critical role in the inflammatory environment, which is consistent with the observation that the expression of the IL-35 receptor chain, IL-12Rβ2, in responder cells requires inflammation-induced stimulation (Presky and others 1996).
In this section, we will discuss what is currently known, whether causative or correlative, regarding the role or impact of IL-35 in 6 autoimmune and inflammatory diseases: type 1 diabetes (T1D), rheumatoid arthritis (RA), multiple sclerosis (MS), systemic lupus erythematosus (SLE), IBD, primary Sjögren syndrome (pSS), and atherosclerosis.
Type 1 diabetes
T1D, also known as juvenile diabetes, is a disease characterized by the autoimmune destruction of pancreatic β-cells, and the disease onset and severity are under the influence of both genetic predispositions and environmental risk factors. The hallmark of T1D is β-cell loss in the islets of Langerhans of the pancreas that leads to insulin deficiency, and its destruction is mediated by diabetogenic T cells (American Diabetes Association 2018). The exact mechanisms that pathogenically activate diabetogenic T cells have not been fully elucidated. However, previous studies have shown that there is a large set of potential disease susceptibility genes that have been associated with immunoregulatory pathways (Chen and others 2018). Non-obese diabetic (NOD) mice develop autoimmune diabetes and have been the primary animal model for genetic, mechanistic, and pre-clinical interventions studies for T1D for more than 30 years (Driver and others 2011).
The importance of IL-35 in autoimmune diabetes was first revealed in transgenic NOD mice with ectopic expression of IL-35 in pancreatic β-cells, which resulted in a considerably delayed onset and long-term protection against autoimmune diabetes (Bettini and others 2012). The reduced diabetes incidence and tissue inflammation were associated with decreased intra-islet infiltration and proliferation of islet resident T cells. Further, a study using an engineered adeno-associated virus vector to drive the overexpression of IL-35 in NOD mice demonstrated that β-cell-specific expression of IL-35 was sufficient to prevent autoimmune diabetes onset (Manzoor and others 2017).
In addition to these observations in the NOD mouse model, several studies suggested there might be a beneficial effect of IL-35 for patients with T1D. Two studies independently reported that patients with T1D have decreased IL-35 in serum compared with healthy individuals (Singh and others 2015, 2019). In addition, a clinical study has shown that the circulating level of IL-35 was correlated with the level of functional pancreatic β-cells, as measured by C-peptide concentration. Interestingly, severe loss of β-cell function was associated with a broad reduction of regulatory immune sub-populations, such as IL-35 producing Tregs, Bregs, and CD8+Foxp3+ cells (Espes and others 2017). Although this warrants further investigation, these observations suggest that IL-35 may be able to limit T1D development by regulating self-reactive diabetogenic T cells.
Rheumatoid arthritis
RA is a systemic autoimmune disease caused by the inflammation and destruction of articular cartilage and bones. The hallmark of RA is the presence of the autoantibodies, such as rheumatoid factors and anti-citrullinated protein antibodies. The experimental collagen-induced arthritis (CIA) model initiated by immunization of mice with type II collagen is one of the most extensively utilized animal models of RA, as it recapitulates many features of RA in humans, including the generation of autoantibodies and the activation of pathogenic T cells (Kobezda and others 2014).
By using this CIA model, numerous studies have indicated a protective role of IL-35 in the pathogenesis of RA (Niedbala and others 2007; Kochetkova and others 2010; Maddaloni and others 2018). For instance, the administration of recombinant IL-35 (rIL-35) Fc fusion protein in arthritis-susceptible DBA/1 mice resulted in a significant reduction in the severity and onset of CIA by limiting IL-17 production (Niedbala and others 2007). This is consistent with the other 2 studies in which exogenous IL-35 alleviated CIA symptoms in C57BL/6 mice by inhibiting proinflammatory cytokines IFN-γ and IL-17 while increasing IL-10 production by CD4+CD39+ Tregs (Kochetkova and others 2010; Maddaloni and others 2018). In addition, IL-35 treatment was shown to promote the apoptosis of fibroblast-like synoviocytes, which are primarily responsible for cartilage damage in arthritis (Li and others 2016). Moreover, the administration of rIL-35 in mice with established CIA halted the further disease progression, suggesting the potential of IL-35 as a therapeutic intervention (Kochetkova and others 2010).
However, conflicting results were reported in which the CIA-induced mice receiving IL-35 encoding plasmids into skeletal muscle showed aggravated clinical manifestation of CIA and altered Treg/Th17 ratio (Thiolat and others 2014). This discrepancy may be ascribed to the differences in the experimental designs, in which the administration of IL-35 at early versus late time points may have resulted in the differential impact on disease progression. Further investigation is needed to address the kinetics of the efficacy of exogenous IL-35.
Given those observations in animal models of RA, there has been increasing interest in investigating the immunosuppressive effect of IL-35 in human RA. Clinical studies have shown that circulating IL-35 was lower in patients with RA compared with healthy controls (Nakano and others 2015; Ning and others 2015). Interestingly, there was an inverse correlation between serum IL-35 concentration and disease severity, implying a protective role of IL-35 in human RA pathogenesis. On the contrary, other studies have reported comparable or increased serum levels of IL-35 in patients with RA compared with healthy individuals (Filkova and others 2015; Yin and others 2016; Li and others 2019; Xie and others 2021). Although more mechanistic studies using human samples are required to address this contradiction, it may be associated with the changes in disease microenvironment based on disease progression.
Importantly, the inflammatory environment can also trigger immunoregulatory mechanisms, such as the recruitment and expansion of activated Tregs, which may lead to an increase in both proinflammatory and inhibitory cytokines temporally. Similar to the conflicting observations in mice, further investigation is warranted to improve our understanding of how the production of IL-35, as well as the abundance and phenotype of responder cells, changes longitudinally throughout the disease progression.
Multiple sclerosis
MS is an inflammatory disease of the central nervous system that is caused by aberrant autoimmune destruction of the myelin sheath surrounding nerve cells, which leads to disruption of the crucial neural network and progressive paralysis (Tsokos 2020). As T cell-mediated destruction of myelin is one of the hallmarks of MS, the mouse model of MS, known as EAE, is induced by the immunization of mice with myelin oligodendrocyte glycopeptide and the administration of pertussis toxin.
Before the discovery of IL-35, a study had demonstrated that p35-deficient mice showed increased disease susceptibility, whereas p40 deficient mice were resistant to EAE (Gran and others 2002). This observation suggested that IL-23, but not IL-12, plays a pathogenic role in EAE, whereas p35 exerts a protective function in an IL-12-independent manner, potentially pointing the figure at IL-35. In support of this possibility, a study using Ebi3 −/− mice showed that Ebi3 suppresses Th17 responses and reduces EAE disease severity (Liu and others 2012). Moreover, a recent study reported that exogenous p35 protein ameliorated EAE by promoting the expansion of IL-35+ Tregs and Bregs (Choi and others 2017). Consistent with these findings, EAE was significantly exacerbated in mice that lack p35 or Ebi3 in B cells. In contrast, mice with p40- or p28-deficient B cells showed normal disease progression, indicating that IL-35, but not IL-12 or IL-27, produced by B cells plays a key role in regulating pathogenic T cell responses (Shen and others 2014). Further, EAE pathogenesis was alleviated when mice were vaccinated with IL-35-expressing DCs (Haller and others 2017), further corroborating that IL-35 plays a protective role in EAE.
In human MS studies, IL-35 level was comparable between healthy controls and patients. However, interferon-β (IFN-β) and methylprednisolone upregulated circulating IL-35 in patients with MS, suggesting that increasing IL-35 production may be one of the mechanisms underlying the efficacy of these therapeutic regimens for MS (Jafarzadeh and others 2015).
Systemic lupus erythematosus
SLE is a multigenic autoimmune disease that impacts multiple systems, leading to the destruction of various tissues, including the kidney, skin, and vasculature. There is great heterogeneity among individuals, even between genetically identical twins, in the disease progression, severity, and the number of organs affected. Extensive studies have demonstrated that both innate and adaptive immune systems are intricately involved in the pathology and disease progression in SLE (Herrada and others 2019). For instance, a downregulated complement system and enhanced TLR signaling, such as TLR7 and TLR9, have been associated with increased disease severity (Herrada and others 2019).
On the other hand, as evident in the massive production of autoreactive antibodies and T cell infiltration in the affected organs and tissues, aberrant activation of the adaptive immune system is playing a detrimental and destructive role in SLE (Tilstra and others 2018; Herrada and others 2019). However, surprisingly, a recent study revealed that murine T cells infiltrating nephritic kidneys isolated from MRL/lpr mice, a model of the most severe and organ-specific form of SLE, showed diminished proliferation, cytokine production, and metabolic capacity. These observations suggest that those kidney-infiltrating T cells are dysfunctional, exhausted T cells, as previously described in cancer and chronic infection (Blank and others 2019), despite the ongoing severe lupus nephritis. As discussed earlier, IL-35 has been shown to induce dysfunctional differentiation in the context of tumor-infiltrating T cells (Sawant and others 2019). Therefore, it is possible that IL-35 also plays a role in SLE, such as by driving the differentiation of exhausted T cells upon infiltration into the inflamed organs to limit the further exacerbation of immunopathology.
Several studies have demonstrated that patients with SLE with ongoing flares have a lower concentration of IL-35 in the blood compared with healthy individuals and patients in a latency phase of SLE (Ouyang and others 2014; He and others 2018; Ye and others 2019). Further, the proportion of IL-35+ Bregs was decreased in patients with SLE (Ye and others 2019). In addition, both serum IL-35 concentration and the frequency of IL-35+ Bregs were negatively correlated with disease severity (Ye and others 2019). Importantly, glucocorticoids, a mainstay of treatment in SLE, increased serum levels of IL-35 in patients who had not received immunomodulating therapies (Ouyang and others 2014). Interestingly, a genome-wide association study (GWAS) screening single nucleotide polymorphisms (SNPs) identified a polymorphism in Ebi3 (rs4740), which was correlated with renal dysfunction and hematological disorder in patients with SLE (Guan and others 2019).
A recent preclinical study utilizing lupus-prone MRL/lpr mice demonstrated that exogenous IL-35 treatment significantly alleviated nephritis, which was associated with the expansion of Tregs and IL-10-producing Bregs (Cai and others 2015). Another study using an IL-35 overexpression plasmid also showed that IL-35 upregulation suppressed proinflammatory cytokine production and alleviated disease severity in MRL/lpr mice (Cai and others 2021). Further, although Rituximab, an anti-CD20 monoclonal antibody to systemically deplete B cells, has been used to treat patients with SLE off-label as well as in numerous clinical trials, the treatment has not reached curative efficacy despite the removal of autoantibody-producing B cells (Alshaiki and others 2018). Although the initial report of IL-35-producing Bregs were identified as CD138hiCD22− mature plasma cells that are known to downregulate CD20 expression (Shen and others 2014), it is possible that the systemic depletion of B cells with Rituximab may have eliminated other non-plasma cell populations of IL-35+ Bregs (Wang and others 2014b), resulting in progressive disease without clinical responses in some patients. More extensive preclinical studies are needed to properly assess the therapeutic potential of IL-35 in SLE.
Inflammatory bowel disease
IBD is a chronic inflammatory disorder that is characterized by massive immune responses within gastrointestinal tracts. Although a significant amount of evidence suggests that IBD is an autoimmune disease, many studies indicate that chronic inflammation is not caused by autoreactive immune cells (Zhang and Li 2014). The pathogenesis of IBD is complex as it is influenced by various factors, including genetic background, environmental factors, and microbiota (Zhang and Li 2014). Although many studies have established the importance of Th17 cells in disease pathogenesis, Tregs are also considered as a fundamental player in IBD, which are involved in the maintenance of homeostasis. Indeed, previous studies have demonstrated that microbiota-induced generation of RORγt+Foxp3+ Tregs in the intestine is critical in maintaining the immune homeostasis in the gastrointestinal tracts (Hepworth and others 2015; Yang and others 2016; Wheaton and others 2017). Although RORγt+Foxp3+ Tregs have been reported to produce an increased level of IL-10 and potently prevent T cell-mediated intestinal inflammation (Yang and others 2016), it is still unclear whether IL-35 is involved in the regulatory mechanisms utilized by RORγt+Foxp3+ Tregs.
Nevertheless, there have been several studies indicating that Treg-derived IL-35 plays a robust immunosuppressive role in IBD. For instance, Ebi3- and p35-deficient Tregs were not sufficient to control T cell-mediated intestinal inflammation (Collison and others 2007). Further, consistent with the previously described “infectious tolerance” orchestrated by Treg-derived IL-35 and differentiated iTr35 cells, iTr35 cells were sufficient to prevent T cell-mediated experimental colitis (Collison and others 2010), demonstrating a potent suppressive role of T cell-derived IL-35. Moreover, an increasing number of studies have shown a beneficial role of rIL-35 in preventing colitis in different IBD mouse models, implying that exogenous IL-35 is a potential therapeutic candidate to prevent the progression of IBD (Wirtz and others 2011; Wang and others 2018, 2019; Zhang and others 2018).
In humans, a recent study reported that serum IL-35 levels were significantly lower in patients with IBD compared with healthy individuals (Li and others 2014). This finding was corroborated by another study in which circulating B cells isolated from Crohn's disease, a relapsing IBD characterized by extraintestinal inflammation throughout the gastrointestinal tract, did not produce IL-35 and failed to upregulate Ebi3 and Il12a transcripts upon stimulation (Zhao and others 2020). However, it remains unclear whether IL-35 production in those patients diminished as a result of the chronic inflammation or the inherent inability to produce IL-35 caused the development of the intestinal inflammation. Thus, further investigation is warranted.
Atherosclerosis
Atherosclerosis is a lipid-driven chronic inflammatory disease that is characterized by inflammation and immune infiltration in the arterial wall that underlies coronary artery disease (Weber and Noels 2011). An increasing amount of evidence suggests that autoinflammation triggered by self-antigens contributes to disease pathogenesis. Apoe −/− mice are a well-established mouse model to study human atherosclerosis, as they also display impaired lipoprotein clearance and develop atherosclerotic lesions (Ye and others 2020). IL-35 levels in Apoe −/− mice with atherosclerosis were significantly higher than wild-type mice (Wang and others 2014a; Li and others 2018). Consistent with these data, a recent study using Ldlr −/− mice showed that mucosal inoculation of type V collagen effectively induced tolerance and ameliorated plaque burden in an IL-35-dependent manner, indicating that IL-35 may be involved in ameliorating atherosclerosis (Park and others 2016).
Moreover, the administration of IL-35 suppresses the development of atherosclerosis in Apoe −/− mice, further corroborating the therapeutic potential of IL-35 against atherosclerosis (Tao and others 2016; Li and others 2018). Consistent with observations in mouse models, clinical data have also indicated a protective role for IL-35 in atherosclerosis. For instance, circulating IL-35 levels are significantly lower in patients with severe disease, such as those with stable angina pectoris, unstable angina pectoris, or acute myocardial infarction (Lin and others 2012). However, the exact mechanism by which IL-35 regulates atherosclerosis remains unclear. Thus, further investigation is warranted to rationally develop a novel IL-35-based therapeutic approach.
Primary Sjögren syndrome
The significance of IL-35 has also been described in other autoimmune diseases, including pSS, which is an autoimmune disorder with dry eyes and dry mouth symptoms. Similar to other autoimmune diseases discussed earlier, the serum level of IL-35 is inversely correlated with disease severity, where active patients with pSS have less circulating IL-35 (Fogel and others 2018; Guo and others 2018; Han and others 2018). In addition, GWAS revealed that an SNP within the Il12a locus (rs485497) was associated with pSS and the serum level of IL-35 was decreased in patients, particularly the ones with lymphoma, implicating a regulatory role for IL-35 in pSS (Lessard and others 2013; Fogel and others 2018).
Overall, the current spectrum of knowledge suggests that IL-35 may curb the development of various inflammatory and autoimmune diseases in experimental models and that IL-35 largely plays a protective role in human autoimmune diseases.
IL-35 in Infectious Diseases
Both innate and adaptive immune systems collaboratively orchestrate robust effector immune responses against invading pathogens, such as bacterial, viral, fungal, and other parasitic pathogens (Parkin and Cohen 2001). However, the resolution of inflammation mediated by regulatory mechanisms is crucial to prevent unwarranted immunopathology. Tregs and immunosuppressive cytokines are key factors in maintaining peripheral tolerance during infection (Ng and others 2013; Stephen-Victor and others 2017). Similar to IL-10, several studies have reported that IL-35 is upregulated under various infectious diseases conditions, including hepatitis (Zhou and others 2015; Liu and others 2017, 2021; Shao and others 2017), influenza (Wang and others 2016a), and sepsis (Cao and others 2015). For instance, influenza A virus infection induces p35 and Ebi3 transcription in both peripheral blood mononuclear cells and lung epithelial cells (Wang and others 2016a). In addition, an elevated serum level of IL-35 was reported in patients with sepsis, which is consistent with increased concentration of IL-35 in the blood and peritoneal lavage from mice with abdominal sepsis, suggesting an important role of IL-35 during infection (Cao and others 2015).
In addition, a clinical study showed that a proportion of peripheral blood isolated from patients infected with hepatitis B virus (HBV) contained IL-35-producing CD4+ T cells, whereas it was undetectable in healthy individuals (Liu and others 2011). In another clinical study, the IL-35 concentration in serum was dramatically increased in patients with HBV compared with healthy controls, and the concentration of IL-35 correlated with viral burden (Zhou and others 2015). Moreover, although the frequency of CD4+ T cells and Tregs positively correlated with serum levels of IL-35, CD8+ cytotoxic T lymphocytes (CTLs) showed an inverse correlation (Zhou and others 2015; Shao and others 2017). These observations suggest that HBV infection results in the upregulation of IL-35 in CD4+ T cells and the expansion of Tregs, affecting the anti-viral responses in CTLs. This is supported by an in vitro study in which IFN-γ production and the expansion of HBV-specific CD8+ T cells from patient blood samples were reduced in the presence of IL-35 during antigen recall responses (Li and others 2015).
In addition, a recent study indicated that IL-35-producing B cells were also enriched in patients with persistent HBV infection, which was negatively correlated with the number of IFN-γ-producing CD4+ and CD8+ cells (Liu and others 2021). Further, a recent study demonstrated that the administration of rIL-35 accelerated viral replication from the blood and liver in an HBV transgenic mouse model, indicating that IL-35 negatively impacts anti-virus responses (Tao and others 2018).
Similarly, IL-35 is also linked in the pathogenesis of chronic hepatitis C virus (HCV) infection in which a positive correlation between serum IL-35 levels and HCV copy number has been reported (Liu and others 2017; Taghinejad and others 2020). Further, IL-35 significantly suppresses IFN-γ, IL-6, IL-8, and TNF-α secretion by HCV-infected Huh7.5 cells via the inhibition of pSTAT1 and pSTAT3 activation (Liu and others 2017).
These observations suggest that IL-35 plays an important immunomodulatory role in the development of hepatitis or potentially other infectious diseases and may serve as a biomarker for disease progression. However, whether the production of IL-35 is induced by the host to maintain equilibrium and avoid inflammation-mediated tissue damage or by virus to facilitate its own replication and survival remains unclear.
Conclusions and Future Directions
Over the past decade or so since the discovery of IL-35, substantial effort has been made to understand the biology of IL-35 and its involvement in different disease environments (Table 1). IL-35 has emerged as not only a potent suppressive cytokine directly limiting the inflammatory response, but also an initiator of “infectious tolerance” in which continuous generation of IL-35-producing iTr35 cells and Bregs establishes a positive feedback loop to amplify this immunoregulatory signal. Therefore, IL-35 represents a great therapeutic target and/or diagnostic biomarker for cancer and autoimmune, inflammatory, and infectious diseases. However, several important gaps in our understanding of IL-35 biology and function need to be filled to facilitate therapeutic development.
How important is IL-35? Several significant challenges have limited a full dissection of this critical question. For instance, the generation of purified, untagged rIL-35 remains a significant challenge limiting functional analysis. Another important unresolved question is how heterodimer stability is maintained and regulated in vivo. With increasing understanding of IL-35 biology, more novel functions of IL-35 beyond the inhibition of T cell proliferation may be uncovered. Whether there are any other functions of IL-35 remains undetermined.
How do different cell populations respond to IL-35 and how is downstream signaling propagated? Given that IL-35 receptor utilization and downstream signaling differs between T cells and B cells, other cell populations may also respond to IL-35 differently. A related obstacle is the lack of a crystal structure of IL-35 with or without its receptors. Further, the lack of efficient antibodies to detect IL-35 and the IL-35 receptors is also a hindrance to advancing our understanding of receptor utilization. In addition, since IL-35 shares receptors with numerous other cytokines, including IL-12 and IL-27, whether IL-27 and IL-35 could compete for gp130 while IL-12 and IL-35 compete for IL-12Rβ2 requires further investigation. The generation of mouse models with cell-type specific deletion of those receptor chains as well as mutated receptors where the binding site is disrupted to allow interaction with only 1 specific cytokine will help improve our understanding of the IL-35 receptor expression patterns as well as mechanisms of receptor signaling and their downstream functional consequences.
Are there any other cellular sources of IL-35? Although IL-35 was initially reported as a Treg-derived inhibitory cytokine, subsequent studies have rapidly expanded the pool of cells that can make IL-35, including Breg, CD8+Foxp3+ Treg, tolDCs, and tumor metastasis-associated macrophages in mice. Thus, whether there are other cell types that expresses high levels of Ebi3 or Il12a transcripts and could also secrete IL-35 remains elusive. Moreover, analogous studies are needed to investigate whether other cellular sources exist that can produce IL-35 in humans.
What is the true contribution of IL-35 in different diseases? Due to the chain sharing feature of the IL-12 family, it has been challenging to distinguish the contributions of IL-35 from IL-12 and IL-27. Despite the emerging studies that investigated the role of IL-35 in numerous disease settings (Table 1), mechanistically driven experiments are lacking that could define exactly how IL-35 regulates disease pathogenesis.
Does the Ebi3-binding repertoire expand beyond p28 and p35? A recent study characterized a novel interaction between Ebi3 and p19, coined IL-39, when co-expressed in activated B cells (Wang and others 2016b). Moreover, monomeric Ebi3 has been reported to facilitate IL-6 trans-signaling via gp130 in the absence of IL-6Rα (Chehboun and others 2017), suggesting there are other additional complexes formed by Ebi3. These observations further increase the complexity of the physiological interpretation of studies with Ebi3- or p35-targeted mouse models. Thus, the generation of other cell-type restricted conditional Ebi3 or/and p35-deficient mice may provide more insight into the IL-35 mechanism. Further, more studies are needed to determine whether there are any other heterodimeric members of the IL-12 family.
In summary, IL-35 plays a critical role in suppressing immune responses and substantial evidence highlights a role for IL-35 in cancer and autoimmune, inflammatory, and infectious diseases in both animal models and human patients. However, further mechanistic dissection of IL-35 biology and function are needed to facilitate its therapeutic targeting in cancer and autoimmune, inflammatory, and infectious diseases.
Footnotes
Authors' Contributions
All authors wrote and edited the article.
Acknowledgments
The authors wish to thank everyone in the Vignali Lab (
Author Disclosure Statement
The authors declare competing interests. D.A.A.V. and C.J.W. have submitted patents covering IL-35 that are pending or approved and are entitled to a share in the net income generated from licensing of these patent rights for commercial development. D.A.A.V.: cofounder and stock holder—Novasenta, Tizona, Trishula, and Potenza; stock holder—Oncorus, Werewolf, Apeximmune; patents licensed and royalties—Novasenta, Astellas, Tizona, BMS; scientific advisory board member—Tizona, Werewolf, F-Star, Bicara; consultant—Astellas/Potenza, BMS, Incyte, Almirall, G1 Therapeutics; research funding—BMS, Astellas/Potenza, Tizona, and Novasenta. All other authors have declared that no competing financial interests exist.
Funding Information
This work was supported by the National Institutes of Health—R01 CA203689 and CA263850.