
Abstract
Dedication: This article is dedicated to Howard Young, an exceptional scientist who has provided outstanding mentorship to many postbaccalaureates, graduate students, and postdoctoral fellows during his career. Howard has been a colleague to many and was never tired of learning new things. He has brought “thinking out of the box” to the level of an art form and has always provided thoughtful and constructive suggestions to those who have sought his counsel. I am personally greatly indebted to Howard for his guidance in molecular biology over the past 30 years, and hope that we will continue to share a passion for learning and mentoring others for years to come. Thank you, Howard! —Stephanie N. Vogel
The SARS-CoV-2 pandemic has led to an unprecedented explosion in studies that have sought to identify key mechanisms that underlie the ravaging aspects of this disease on individuals. SARS-CoV-2 virus gains access to cells by (1) binding of the viral spike (S) protein to cell-associated angiotensin-converting enzyme 2 (ACE2), a key receptor in the renin–angiotensin system (RAS), followed by (2) cleavage of S protein by a cellular serine protease (“S protein priming”) to facilitate viral entry. Dysregulation of the RAS system has been implicated in the spectrum of clinical symptoms associated with SARS-CoV-2, including hypercytokinemia, elevated markers of endothelial injury and thrombosis, and both localized and systemic inflammation. However, the underlying mechanisms have yet to be fully delineated.
Over the past decade, we have documented that acute lung injury (ALI) induced by experimental influenza infection is toll-like receptor 4 (TLR4) dependent, and mediated by a host-derived “Danger Associated Molecular Pattern (DAMP),” High Mobility Group Box-1 (HMGB1) [rev. in Shirey et al (2021)]. Therapeutic targeting of either TLR4 signaling or HMGB1-induced TLR4 signaling (by small molecule inhibition of the TLR4 coreceptor, MD-2 [Shirey et al, 2016, 2021]) protected against influenza-induced hypercytokinemia and ALI in rodent models. In the following article, we provide evidence for potential cross-talk between the renin–angiotensin system (RAS) pathway and innate immune signaling through TLR4 that underlies ALI.
The RAS and Its Role in ALI/Acute Respiratory Distress Syndrome
A complex multicomponent signaling system, RAS (Fig. 1), has been recognized as central in ALI in multiple experimental models, that is, acid aspiration, lipopolysaccharide (LPS)-induced ALI, sepsis-induced ALI, pancreatitis, trauma, and respiratory bacterial and virus infections including anthrax, SARS CoV, SARS-CoV-2, and influenza (eg, H7N9, H5N1) [rev. in Imai et al (2008); Yang et al (2014)]. In brief, angiotensin I (Ang I) is a decapeptide formed by enzymatic cleavage of a precursor molecule, angiotensinogen, by renin.

Model for RAS illustrating opposing pro- and anti-inflammatory signaling pathways induced by ACE and ACE2, respectively. ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; ALI, acute lung injury; ARDS, acute respiratory distress syndrome; RAS, renin–angiotensin system.
Ang I is then cleaved by a transmembrane-associated enzyme, angiotensin-converting enzyme (ACE), into an octapeptide, Ang II, that interacts with its receptor, AT1R, to cause vasoconstriction that, under nonphysiological conditions leads to an excessive increase in blood pressure, dysregulated vascular permeability, and edema. A second closely related transmembrane-associated enzyme, ACE2, cleaves Ang I to the nanopeptide Ang(1–9) and Ang II to the heptapeptide Ang(1–7), which act on 2 other receptors, AT2R and Mas, respectively, to initiate the vasodilatory pathway, thereby counteracting the excessive vasopressor ACE axis of the RAS (Fig. 1).
Thus, ACE inhibitors (ACEi) and AT1R antagonists (ARBs) are widely used to control hypertension (Goodman and Gilman, 2018). Overexpression of ACE2 is protective in models of myocardial infarction, whereas ACE2-deficient mice are more susceptible to heart failure induced by cardiac banding [rev. in Imai et al (2008); Luft (2016)] and exhibit increased vascular permeability, neutrophil (PMN) accumulation, pulmonary edema, and worsened lung function in experimental models of acute respiratory distress syndrome (ARDS) [eg, acid aspiration, LPS induced, and sepsis induced; Imai et al (2008); Wiese et al (2021); Yang et al (2014)]. Blocking RAS with ACEi or ARBs or treatment with catalytically active ACE2 attenuates influenza-induced ARDS in mice (Yang et al, 2014).
In a large retrospective analysis, it was found that persons taking ACEi or ARBs exhibited a lower incidence of influenza A infection (Chung et al, 2020). Thus, the ACE/AngII/AT1R axis is counteracted by the ACE2/Ang(1-7)/Mas and ACE2/Ang(1-9)/AT2R axes: increased ACE2 correlates with decreased Ang II, and is an indicator of organ protection (decreased pulmonary vasoconstriction, blood pressure, atherosclerosis) and protects against pulmonary angioedema. Conversely, decreased ACE2 increases angioedema, pulmonary vascular permeability, edema, and ARDS (Zanorano Cuervo and Grandvaux, 2020). AT1R activation also leads to proinflammatory signaling.
That the spike (S) proteins of SARS viruses enable infection by binding to human ACE2 (hACE2) (Wrapp et al, 2020) followed by cleavage of S protein by a cellular protease to facilitate viral entry (Hoffmann et al, 2020), has led to intensive investigation of the role of the RAS pathway in ALI/ARDS in COVID-19 patients. ACE2 is highly expressed in the lung on tissue macrophages, alveolar epithelial and endothelial cells, as well as in the heart, kidney, and gastrointestinal tract (Song et al, 2020). Binding of S to hACE2 results in its depletion by internalization or shedding, leading to Ang II accumulation and the deleterious effects of excess AT1R signaling.
Ang II also upregulates TLR4 expression (Nair et al, 2015), stimulates reactive oxygen species (ROS), and activates NF-κB and PI3K through AT1R, resulting in release of proinflammatory cytokines, chemokines, and adherence molecules. Conversely, ACE2-derived Ang(1–7) delivers an anti-inflammatory signal through Mas (Iwasaki et al, 2021). Exogenous Ang(1–7) inhibits interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) production by LPS-stimulated macrophages (Iwasaki et al, 2021), suggesting interaction between the RAS and TLR4/MD-2 signaling pathways.
In contrast to SARS viruses, for which infection is ACE2 dependent, the role of the RAS in ALI/ARDS is less well understood for agents that do not require RAS components for entry into cells. Like SARS viruses, influenza H5N1 infection of mice downregulates ACE2 expression and increases serum Ang II levels, as seen in H7N9-infected patients (Yang et al, 2014; Zou et al, 2014), yet the mechanism is controversial. ACE2−/− mice exhibited greater ALI in response to H5N1 infection than WT mice (Zou et al, 2014). Sfera et al (2020) proposed that “endogenous Ang II toxicity” is a “common pathophysiological denominator” for induction of ARDS mediated by premature vascular senescence, dysfunctional coagulation (as evidenced by increased levels of D-dimer), and dysregulated immunity.
Excess Ang II was proposed to act as a “mitochondrial toxin,” leading to increased ROS and IL-6. Melo et al (2020) showed that treatment of WT mice, but not Mas−/− mice, with Ang(1–7) on days 3–10 after influenza infection led to decreased lethality, decreased infiltrating leukocytes in airways, less myeloperoxidase in lungs, and decreased viral titers. Our data extend these findings: infection of WT mice with mouse-adapted influenza strain A/PR/8/34 (PR8) resulted in a time-dependent decrease in Ace2 mRNA expression, whereas Tnf mRNA (and other cytokine mRNAs; data not shown) increased over the same time (Fig. 2a).
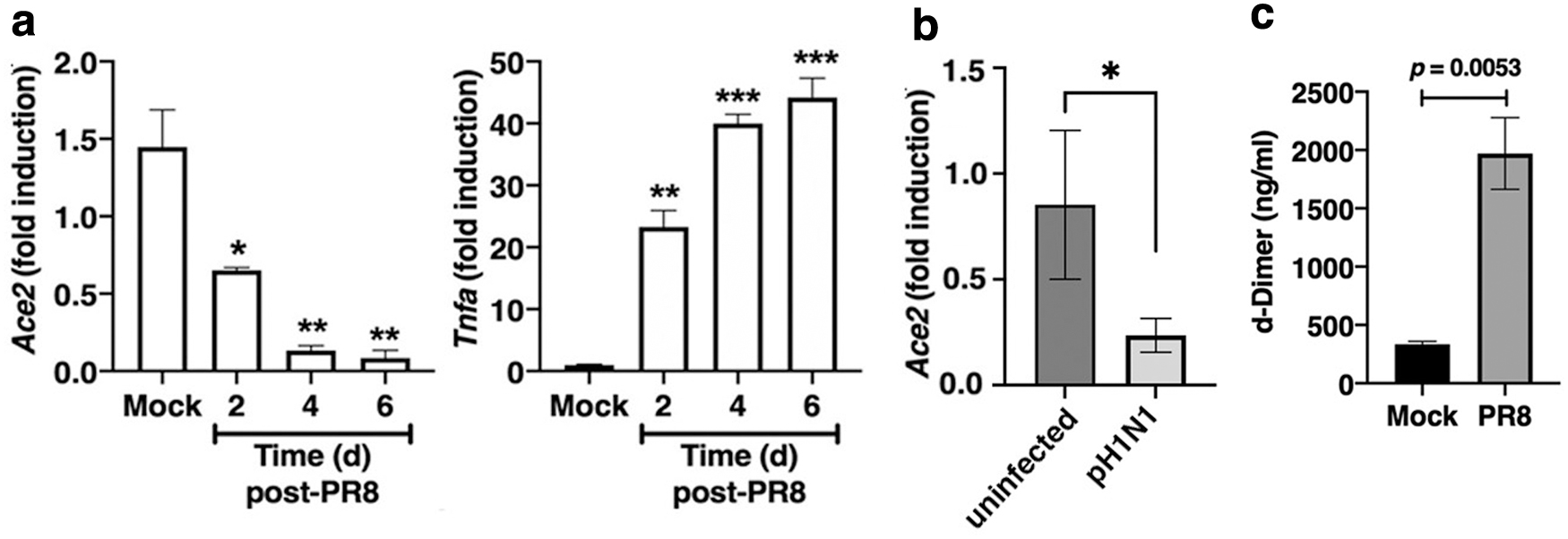
Downregulation of Ace2 mRNA was also observed in cotton rats, a rodent species, Sigmodon hispidus, that is uniquely susceptible to nonadapted human viruses, when infected with human pH1N1 (Fig. 2b). Furthermore, in another similarity to SARS-CoV-2, we also observed that PR8 infection of WT mice resulted in a significant increase in D-dimer, a breakdown product of blood clotting, in the sera from infected mice (Fig. 2c).
These data support the possibility that influenza downregulates lung ACE2 expression at the level of transcription, in contrast to S protein that downregulates ACE2 by internalization or enzyme-mediated shedding. However, Liu et al (2014) reported that H1N1 infection of ACE2-expressing transfectants decreased ACE2 protein expression due to proteosome degradation, indicating that multiple inhibitory checkpoints could result in the inhibition of ACE-2 expression by influenza.
Association of RAS with HMGB1/TLR4/MD-2-Mediated Signaling
Recent studies suggest an interplay between TLR4 and RAS signaling pathways. Nair et al (2015) reported that TLR4/MD-2 signaling induced by HMGB1 resulted in Ang II-induced renal inflammation. Ang II increased TLR4 expression and release of HMGB1 and inflammatory cytokines in rat epithelial cells that were blocked by the AT1R ARB losartan, TLR4 siRNA, or anti-HMGB1 antibody. The authors concluded that HMGB1-induced TLR4 signaling contributes to Ang II-mediated inflammation. Furthermore, TLR4-deficient mice were resistant to LPS-induced acute renal injury (Cunningham et al, 2004).
Nair et al (2015) proposed that Ang II upregulates NF-κB that elicits cytokines and chemokines, ROS, and release of HMGB1 that, in turn, acts on TLR4/MD-2 to increase cytokine and ROS production, leading to hypertensive renal dysfunction. Pretreatment of human epithelial cells with the ACE2 activator, diminazene aceturate (DIZE) (Gjymishka et al, 2010), increased ACE2 expression; blunted LPS-induced IL-6, IL-8, and MCP-1 mRNA and protein; and inhibited LPS-induced AT1R mRNA expression (Tao et al, 2016).
In patients with stroke, polycystic disease, diabetic neuropathy (DN), or myocardial infarction, or in a model of hypoxia, HMGB1 and cytokine levels are increased when ATR1 is activated or when ACE2-mediated signaling is blocked (Feng et al, 2020; Luft, 2016; Qi et al, 2016). Ang II-activated macrophages exhibit an “M1” phenotype with increased levels of both HMGB1 and proinflammatory cytokines (Jeong et al, 2019; Zhou et al, 2018). Activation of the ACE2-Ang(1–7)-Mas pathway in a model of LPS-induced ALI inhibited TLR4/MD-2-induced signaling and alleviated inflammatory cell infiltrate, permeability, and edema.
Conversely, antagonizing Ang(1–7) or Mas attenuated the protective effect of ACE2 activation (Ye and Liu, 2019). Finally, Ang II was shown to stimulate PKC, PTK, MAPK, ERK, JNK, p38, and AP1, and promote oxidative stress and reduced nicotinamide adenine dinucleotide phosphate oxidase, leading to enhanced NF-κB and cytokine production. TLR4 is also upregulated in renal cells in DN, and TLR4−/− mice were protected from diabetic neuropathy. The authors concluded that blocking TLR4/MD2 attenuates DN by reducing renal RAS activation. Although Xu et al (2017) reported that Ang II binds MD-2 and stimulates TLR4, Okechukwu et al (2021) failed to confirm this finding. Collectively, these studies indicate that Ang II activation of AT1R leads to HMGB1 release that, in turn, stimulates inflammation through TLR4/MD-2.
Gene Expression in a Human Macrophage Cell Line Stimulated with LPS or SARS-CoV-2 S Protein
The previous section suggests potentially important interactions between the RAS and TLR4/MD-2 signaling pathways; however, recent studies (in silico analyses and direct binding studies) suggest that SARS-CoV-2 S protein may interact directly with TLR4 or the TLR4/MD-2 complex to induce signaling (Battacharya et al, 2020; Choudhury and Mukherjee, 2020; Zhao et al, 2021). Zhao et al (2021) reported that SARS-CoV-2 stimulated induction of IL-1β mRNA in the human THP-1 macrophage-like cell line. In addition, recombinant trimeric S protein induced gene expression in a dose-dependent manner that was inhibited by the TLR4 antagonist Resatovid (TAK-242).
Of note, we previously reported that TAK-242, which blocks LPS signaling by interacting with the intracellular Toll-IL-1 receptor resistance domain of TLR4, also blocked influenza-induced ALI in mice (Shirey et al, 2020). In addition to THP-1 cells, Zhao et al (2021) reported that S protein induced gene expression in murine bone marrow-derived macrophages from wild-type mice, but not from TLR4−/− mice. Inhibition of the TLR4 coreceptors, MD-2 and CD14, also decreased IL-1β mRNA expression, whereas TLR2 deficiency had no effect. RNAseq was performed and they reported that S protein upregulated expression of genes encoding interleukins, chemokines, and interferon-stimulated response genes. Induction of macrophage gene expression was not dependent on ACE2 or the protease TMPRSS2 that are required for viral entry into cells.
Independently, we characterized gene expression induced by S protein in the THP-1 cell line. Initially, we compared induction of a handful of highly LPS-inducible genes (MyD88- and TRIF-dependent) by qRT-PCR in response to purified recombinant trimeric S protein (expressed in a mammalian expression system (Stadlbauer et al, 2020). Highly purified Escherichia coli K235 LPS (1 ng/mL) induced each of these genes over a 6 h time course, but only those genes in panel “a” were induced by S protein (3 μg/mL), and to a much lesser extent than by LPS (Fig. 3).

Human THP-1 cells (ATCC) were differentiated into macrophages with PMA then stimulated with media only, LPS (1 ng/mL; solid circles), or purified S protein (3 μg/mL; open squares) based on preliminary studies to insure optimal gene induction. At each time point, mRNA for the indicated human genes was analyzed by qRT-PCR. Data derived from 2 independent experiments.
In these same samples, ACE2 mRNA was not increased over this 6-h time course by either LPS or S protein (33 ± 1.3 cycles). In addition, treatment of THP-1 cells with the ACE-2 activator, DIZE, resulted in gene induction patterns that were similar to those induced by recombinant S protein in Fig. 3 (data not shown).
This is in contrast to the observation by Zhao et al (2021) that LPS and S protein induced gene expression to similar extents. Our data from this small panel of genes also revealed that S protein-inducible genes could not be distinguished based on their dependence upon MyD88 signaling or TRIF signaling (ie, CXCL10, IFNB1 are TRIF dependent). Interestingly, all S-inducible genes in this small panel were chemokine genes, suggesting that infiltrating neutrophils and monocytes likely mediate the cytokine storm associated with ALI. The recombinant S protein prepared in our laboratory was unlikely to be contaminated by LPS based on a limulus amoebocyte lysate (LAL) assay (<0.01 ng/μg endotoxin) and by mass spectrometry, as well as the observation that S protein failed to induce the LPS-inducible genes in Fig. 3b.
In contrast to Zhao et al (2021), our S protein preparations failed to activate murine macrophages, and eritoran, a TLR4 antagonist that acts by competitive inhibition of the LPS binding site on MD-2 (Park et al, 2012), blocked LPS-induced gene expression but failed to block S protein-induced CXCL2 mRNA in THP-1 cells (Fig. 4). Although this suggests that S protein does not bind to the LPS-binding site on MD-2, it does not preclude the possibility that S protein either binds to a different site on MD-2 or to TLR4 itself, as has been suggested by in silico modeling (Battacharya et al, 2020; Choudhury and Mukherjee, 2020).
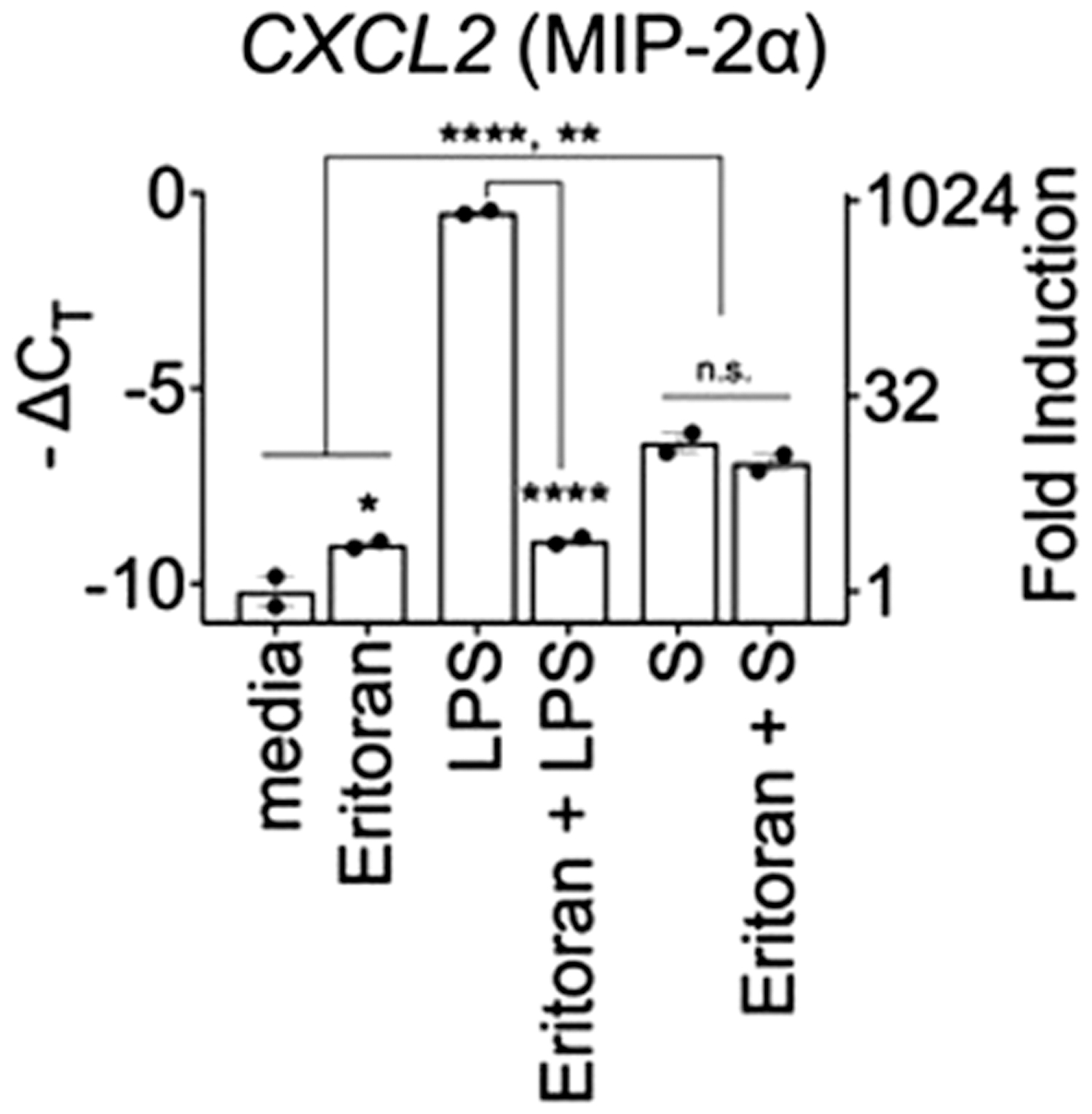
Human THP-1 cells (ATCC) were differentiated into macrophages with PMA then pretreated with eritoran (1 μg/mL, 30 min) followed by stimulation with media only, LPS (1 ng/mL), or purified S protein (3 μg/mL) for 4 h. CXCL2 mRNA was analyzed by qRT-PCR. HPRT was used to normalize for gene expression. Data represent 1 of 2 independent experiments with similar outcomes. *P < 0.05; **P < 0.01; ****P < 0.0001.
To extend these findings, whole transcriptome microarray analyses (Clariom D, Affymetrix; 2 independent experiments) were carried out on THP-1 cells stimulated with S protein or LPS for 4 h, based on optimal gene induction observed in Fig. 3a: 1,361 transcripts were regulated by stimulation with S protein and 9,279 transcripts with LPS. After exclusion of noncoding and unassigned transcripts, 98 genes were responsive to S protein, but not LPS (“S-unique”), and 3,630 genes were LPS responsive, but not S responsive; an LPS- and S-responsive subset comprised 91 genes (“shared”). Ingenuity pathway analysis (QIAGEN IPA) identified VDR/RXR signaling as the top canonical pathway unique to S protein stimulation.
Analysis of “shared” genes confirmed that transcriptional responses to S were consistently less robust than those induced by LPS; however, many antiviral genes (eg, IFIT1, IFIT2, MX1, DDX58) topped this list, and some of the chemokine genes upregulated by S (Fig. 3a) were confirmed. Taken collectively, these findings further support a possible interplay between TLR4/MD-2 and RAS signaling pathways. Additional studies will be required to directly evaluate the role for TLR4/MD-2 signaling in RAS activation and vice versa. However, the data cited and presented herein strongly support the hypothesis that these 2 signaling pathways are integrally linked and impact each other.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This article was supported, in part, by NIH AI125215 (S.N.V./J.C.G.B.), AI123371 (S.N.V.), and AI109926 (J.C.G.B.).