
Abstract
Type III interferon, or interferon lambda (IFNλ), was discovered 20 years ago and has been studied primarily for its role in combatting viral infections. However, it is also induced in response to certain bacterial infections but its roles and effects in this context are relatively poorly understood. In this mini review, we discuss the roles of IFNλ signaling in bacterial infections, highlighting its deleterious or protective effects for different infections. We also discuss a couple of recent studies showing that some bacteria possess defense mechanisms against the effects of IFNλ. We hope that this review will spur further investigation into the roles of IFNλ in the context of bacterial infections and will promote considerations of its therapeutic potential for these infections.
Introduction
Interferons (IFNs) are cytokines that play a critical role in the innate immunological response to viruses and other pathogens, as well as in the generation of adaptive immune responses (Pestka et al., 2004). They are classified into 3 families: type I, type II, and type III. The type I family comprises 17 members in humans and 18 in mice, with IFNα and IFNβ being the most well studied (Pestka et al., 2004). The type II family consists of IFNγ (Schroder et al., 2004) and the type III family consists of IFNλ (Ye et al., 2019). There are 4 known isoforms of IFNλ in humans (IFNλ1, IFNλ2, IFNλ3, and IFNλ4) (Kotenko et al., 2003; Prokunina-Olsson et al., 2013) and only 2 isoforms in mice (IFNλ2 and IFNλ3), which are functionally somewhat redundant (Lasfar et al., 2006; Rich et al., 2021).
Many of the publications cited in this short review used mouse studies; therefore, the isoform of IFNλ is not specified. In the cases where human cells or patient samples were studied, we highlight the isoforms being investigated in the study. Each family of IFNs signals by binding its respective receptor on the cell surface, activating JAK tyrosine kinases associated with the cytoplasmic domains of the receptors. The activated JAKs phosphorylate tyrosine residues on the receptor that serve as binding sites for STAT proteins that are recruited, dimerize, and then translocate to the nucleus and promote transcription of interferon-stimulated genes (ISGs) (Mesev et al., 2019; Schroder et al., 2004).
Since the discovery of the type III IFN family (IFNλ) in 2003 (Kotenko et al., 2003; Sheppard et al., 2003), much of the work has been focused on the biological roles and responses to viral stimuli (Hemann et al., 2017; Hermant and Michiels, 2014). Recent reviews have highlighted its antiviral roles in respiratory infections (Lozhkov et al., 2020), in hepatitis B infection (Novotny et al., 2021) and its therapeutic potential for other viral infections (Lasfar et al., 2016). A few other reviews have considered the role of IFNλ signaling in bacterial infections (Alphonse et al., 2022a; Cohen and Parker, 2016; Kotenko et al., 2019).
This mini review will discuss the roles of IFNλ signaling in the context of bacterial infections, contrast deleterious and beneficial effects of IFNλ, and highlight some of the major questions currently in the field. Although IFNλ is known to have potent activity against certain viral infections, its roles in response to bacterial infections are not well understood and appear to be quite complex. IFNλ expression is upregulated in response to several bacterial infections (as described in the next section). However, in certain bacterial infections IFNλ signaling appears to be deleterious and in others protective.
Induction of IFNλ by Bacteria (and Bacterial Ligands)
Several reports demonstrate the upregulation of IFNλ by bacteria or bacterial ligands. One study found that the intracellular bacterial pathogen Listeria monocytogenes induced IFNλ expression in infected cultured human epithelial cells, although the induction mechanism was not identified (Lebreton et al., 2011). Interestingly, they found that the secreted L. monocytogenes virulence factor LntA targeted the chromatin repressor BAHD1 in the host cell nucleus, relieving its repression of downstream ISGs and modulating the IFNλ-mediated immune response to bacterial infection.
The same group also found that IFNλ expression was upregulated in cultured human epithelial cells infected with other Gram-positive bacteria, such as Staphylococcus aureus, Staphylococcus epidermidis, Enterococcus faecalis, and Mycobacterium tuberculosis, whereas the Gram-negative bacteria Salmonella enterica serovar Typhimurium, Shigella flexneri, and Chlamydia trachomatis did not, leading them to speculate that IFNλ may be an important modulator of responses to Gram-positive, but not Gram-negative, intracellular bacteria (Bierne et al., 2012).
Another study also observed upregulation of IFNλ expression in murine bone marrow-derived dendritic cells infected with a wide range of S. aureus clinical isolates (Peignier et al., 2020). In contrast to the previous findings that Gram-positive but not Gram-negative bacteria induced IFNλ expression, Cohen and Prince (2013) found that Pseudomonas aeruginosa induced significant production of IFNλ in the lungs in a mouse model of infection. In addition, Odendall et al. (2017) found that Salmonella and Shigella upregulated IFNλ1 and IFNλ2 expression in cultured human epithelial cells and in human or mouse myeloid cells. This response was not dependent on internalization of the bacteria but was induced through Toll-like receptor (TLR) signaling after binding of bacterial ligands at the cell surface.
In addition, Ahn et al. (2019) found that IFNλ expression was induced in a mouse model of Klebsiella pneumoniae respiratory infection and Akoolo et al. (2022) observed increased IFNλ production from bone marrow-derived macrophages incubated with Acinetobacter baumannii. Our group found that IFNλ was also upregulated in the lungs of adult mice infected with the Gram-negative bacterial pathogen Bordetella pertussis (Ardanuy et al., 2020) and this induction was primarily mediated by TLR9 signaling (Ardanuy and Carbonetti, unpublished data).
Beyond cultured cells and mouse models, IFNλ1 and IFNλ4 expression was detected in granulomas in nonhuman primates infected with M. tuberculosis (Talukdar et al., 2022) and IFNλ2 in the sputum of human patients with active pulmonary tuberculosis (Travar et al., 2014). Therefore, both Gram-positive and Gram-negative bacterial infections (and their ligands) can stimulate IFNλ responses. Investigations into the specific bacterial ligands that stimulate these responses have not been reported, and it remains possible that in some infections the stimulating ligand is a host-derived molecule, possibly a damage-associated molecular pattern, released after pathological changes, which can signal through a pattern recognition receptor known to induce IFNλ responses.
Interestingly, some studies have shown that bacterial species among the microbiota can affect induction of IFNλ, although this effect may result in diverse outcomes with respect to viral infections. Jounai et al. (2012) found that strains of Lactococcus lactis induced IFNλ expression in cultured plasmacytoid dendritic cells. Groeger et al. (2020) found that intranasal administration of Bifidobacterium longum or its isolated cell wall could protect mice against influenza virus infection, through an effect that included increased levels of IFNλ, although an IFNλ response just to the bacteria was not investigated. A recent study showed that bacterial microbiota stimulate a homeostatic IFNλ response specifically in epithelial cells in localized patches of the intestinal tract, which protects against enteric virus infection (Van Winkle et al., 2022).
In contrast, Baldridge et al. (2015) found that resident bacteria of the intestinal tract inhibited antiviral effects of IFNλ that resulted in norovirus persistence. In addition, Rodriguez-Hernandez et al. (2021) found that the oral pathogen Porphyromonas gingivalis abolished IFNλ production in response to virus infection, by downregulating multiple genes involved in the antiviral IFN response, whereas other oral bacteria had no effect or increased IFNλ production. Collectively these studies demonstrate the complex interplay between bacteria, IFNλ responses and viral infections, and the diverse effects that can result, presumably because of context-specific factors. We will address this topic further as follows.
Deleterious Effects of IFNλ in Bacterial Infections
Recent studies indicate that IFNλ signaling is deleterious in certain acute bacterial respiratory infection models, including P. aeruginosa, S. aureus, and K. pneumoniae. The impact of IFNλ signaling on these specific bacterial pathogens has been investigated in IFNλ receptor (IFNLR1)-deficient adult mice, where each study found that in the absence of IFNλ signaling, there was reduced pulmonary pathology associated with infection and increased bacterial clearance (Ahn et al., 2019; Cohen and Prince, 2013; Pires and Parker, 2018), indicating that IFNλ signaling is deleterious in these interactions. IFNλ signaling also caused greater lung pathology in B. pertussis-infected mice; however, in contrast to the other infections, IFNλ signaling did not affect bacterial burden (Ardanuy et al., 2020).
The mechanisms leading to IFNλ-mediated increase in pulmonary pathology are still largely unclear. In the context of P. aeruginosa and S. aureus infections, IFNλ inhibited expression of the micro-RNA miR-21, resulting in upregulation of the tumor suppressor PDCD4 in human epithelial cells (Cohen and Prince, 2013). Mice lacking PDCD4 had reduced pulmonary pathology and increased bacterial clearance, demonstrating a role for miR-21 and PDCD4, known modulators of inflammatory signaling, in the effects of IFNλ in these infections.
In lung infection of mouse models with B. pertussis, the specific mechanisms by which IFNλ increases lung inflammatory pathology are still under investigation (Ardanuy et al., 2020). Interestingly, this study showcased an interplay between type I and type III IFNs that has not been observed (to our knowledge) in other bacterial infection models. When IFNλ signaling was absent, infected adult mice did not significantly upregulate type I IFN expression, leading to the possibility that IFNλ increases inflammation through enhancing type I IFN signaling. However, when type I IFN signaling was absent [during infection of IFNα receptor (IFNAR1)-deficient mice], IFNλ expression was upregulated to a greater extent than in infected wild-type mice and this higher level of IFNλ caused increased pulmonary pathology, indicating that IFNλ effects independent of type I IFN signaling can enhance inflammation (Ardanuy et al., 2020).
Although the mechanism of action for promoting pathogenesis is poorly understood for pathogens such as B. pertussis, we are beginning to understand the role that IFNλ signaling plays in promoting pathogenesis in infections with S. aureus and K. pneumoniae. In S. aureus infection, Pires and Parker (2018) demonstrated the importance of IFNλ-stimulated interleukin-1 beta (IL-1β) production by neutrophils and its contribution to lung inflammatory pathology. They noted that IFNLR1-deficient mice had reduced IL-1β production by neutrophils and concomitant lower bacterial loads and reduced lung pathology compared with infected wild-type mice.
When IFNLR1-deficient animals were treated with mouse recombinant IL-1β, the bacterial load increased and lung immunopathology worsened. This demonstrates that a mechanism by which IFNλ can mediate deleterious effects is through IL-1β production by neutrophils. Whether this is a direct effect of IFNλ on neutrophils is unclear, although this is possible since neutrophils express the IFNλ receptor (Linden et al., 2020). The use of neutrophil-specific IFNLR1 knockout mice will be necessary to determine this. In addition, investigation of the pathways by which IFNλ signaling can result in increased production of IL-1β will be informative for the field.
Although IFNλ effects on neutrophils appear to be a significant contributor to pathogenesis in these bacterial infection models, the context-specific nature of this activity is highlighted by the contrasting protective effects of IFNλ signaling on neutrophils in its antifungal properties in a mouse model of Aspergillus infection (Espinosa et al., 2017). This study showed that CCR2+ monocytes produce type I IFNs in response to infection that induce IFNλ production, which in turn acts on neutrophils to promote their antifungal response. This is another example of the interplay between type I and III IFNs in determining outcomes of infection. It also highlights the discrepancies in effects of IFNλ signaling between different infections and the need to study effects in the context of the particular infection model.
Another mechanism by which IFNλ signaling appears to contribute to pathogenesis in bacterial infections is through effects on epithelial barrier integrity. In the case of K. pneumoniae infection, evidence indicates that IFNλ signaling decreases epithelial barrier integrity to facilitate immune cell recruitment but with concomitant bacterial dissemination from the airways (Ahn et al., 2019). In wild-type mice infected with K. pneumoniae, downregulation of lung epithelial tight junction protein expression was observed. In contrast, IFNLR1-deficient mice intranasally infected with K. pneumoniae were significantly protected from bacteremia.
They found that cultured human airway epithelial cells significantly induced IFNλ2/3 in the presence of K. pneumoniae and treatment with IFNλ resulted in transcriptional downregulation of tight junction proteins and decreased barrier integrity. Treatment with IFNλ also increased neutrophil transmigration across the monolayer, whereas treatment with IL-22, a cytokine associated with epithelial recovery, decreased neutrophil transmigration. Therefore, effects on airway epithelial barrier integrity, as well as neutrophil recruitment and activity, appear to be a mechanism by which IFNλ plays a deleterious pro-inflammatory role in certain infections.
IFNλ deleterious effects on epithelial barrier integrity are not unique to bacterial infections. Broggi et al. (2020a) demonstrated that treatment of mice with poly I:C (a surrogate for viral infection) and resulting elevated expression of IFNλ caused greater barrier damage and 100% lethality within 48 h of a subsequent S. aureus respiratory infection. Poly I:C-treated and infected IFNLR1-deficient mice were completely protected from lethality and had significantly reduced lung permeability, confirming the important role of IFNλ signaling in these barrier effects.
Similarly, Major et al. (2020) found that influenza virus-infected IFNLR1-deficient mice had superior epithelial barrier function and lower levels of inflammation than infected wild-type mice. They also found that IFNλ signaling relatively late (11 days) after influenza virus infection resulted in induction of p53 and inhibition of epithelial proliferation and repair. Interestingly, lethally irradiated IFNLR1-deficient mice that were reconstituted with bone marrow cells from wild-type mice were less susceptible to lethal bacterial superinfection post-influenza infection than control wild-type mice, demonstrating that IFNλ signaling in epithelial cells is the pathogenic activity in this scenario.
This theme of IFNλ-mediated reduction of epithelial barrier integrity leading to bacterial superinfection after influenza virus infection has been observed in other studies. Planet et al. (2016) used an influenza virus and S. aureus superinfection model where they demonstrated that IFNLR1-deficient mice were protected from subsequent nasal colonization by S. aureus after virus infection. IFNLR1-deficient mice had increased nasal expression of the barrier-enhancing cytokine IL-22, as well as increased expression of cytoskeletal and structural proteins that enhance barrier function and of antimicrobial peptides.
They also observed IFNλ-mediated effects on the upper airway microbiome that could also affect S. aureus nasal colonization during superinfection. These findings once again point to an important role for IFNλ in reducing protective barrier effects that would enhance bacterial pathogenesis. Additional effects of IFNλ in the context of bacterial superinfection after influenza were reported by Rich et al. (2019), who found that mice treated with adenoviral vectors to overexpress IFNλ during influenza virus infection exhibited increased bacterial loads when superinfected with either S. pneumoniae or S. aureus. Despite no changes in overall immune cell responses such as adhesion molecule expression, reactive oxygen species production and activity, or antimicrobial peptide production, neutrophils showed reduced ability to phagocytose and clear the bacterial superinfection.
The same group also found that IFNλ signaling tracked specifically with the influenza virus infection in mouse models of influenza/bacterial superinfections and that the determining factor for superinfection susceptibility was the level of IFNλ response to influenza virus and not changes to cellular inflammation or viral load (Rich et al., 2021). Therefore, epithelial barrier and neutrophils, cells which are known to express IFNLR1, are once again implicated in the pathogenesis of these infections. It will be important to dissect the molecular events after IFNλ signaling in these cell types, especially in neutrophils, to account for observations of reduced antibacterial activity but increased antifungal activity.
Protective Effects of IFNλ in Bacterial Infections
There are few examples of protective roles of IFNλ signaling in bacterial infections, but they appear to be related to its ability to affect barrier function at mucosal surfaces. As a part of their pathogenesis, many enteric pathogens disrupt the epithelial barrier to enable their spread to underlying tissue. In models where IFNλ signaling was found to be protective, its role was to reinforce intestinal barrier function. For polarized T84 (gut epithelial) cells, IFNλ signaling alone was sufficient to increase the intrinsic transepithelial electrical resistance of the cell monolayer and also prevent barrier damage induced by pathogenic S. flexneri and S. enterica serovar Typhimurium (Odendall et al., 2017).
IFNλ1 treatment of T84 cells also significantly reduced the intracellular replication of Shigella, but there was no difference in Shigella numbers in wild-type or IFNLR1-deficient cells, indicating that cells other than epithelial cells produce IFNλ in this interaction (Alphonse et al., 2022b). Although the molecular mechanism of increased barrier function is poorly understood, some hints may be found in an intestinal ischemia/reperfusion model. Li et al. (2021) found that intracellular tight junction proteins such as claudin-1 may be regulated by IFNλ signaling. In their study, adult mice were randomly assigned to 3 groups: sham, ischemia/reperfusion, and ischemia/reperfusion with treatment of recombinant IFNλ. Six hours after treatment, the mice were euthanized and mucosal permeability was measured and histology analysis performed.
Mice in the IFNλ treatment group had lower permeability and less histology than mice in the ischemia/reperfusion group alone, demonstrating the protective effects of IFNλ on the intestinal epithelia. Of the tight junction proteins assayed, only the expression of claudin-1 was significantly increased in the IFNλ-treated group compared with the ischemia/reperfusion group alone. In addition, treatment of Caco-2 cells, an intestinal cancer cell line, with fludarabine, a pSTAT1 inhibitor, caused a decrease in claudin-1 expression even after IFNλ treatment, whereas other tight junction proteins such as zonula occludens-1 (ZO-1) and occludin did not change, demonstrating the role of IFNλ signaling through STAT1 in the barrier protection activity (Li et al., 2021). Whether or not these observations in cell culture extend to in vivo models of bacterial infection remains to be determined.
One intriguing point that arises from these observations is the discrepancy in the effects of IFNλ signaling on barrier integrity during bacterial infections between the respiratory and gastrointestinal tracts. Although based on a limited number of studies, the data suggest a largely harmful effect of IFNλ signaling in the respiratory tract but a protective effect in the gastrointestinal tract. In the context of virus infections, best studied for influenza, IFNλ signaling is protective in the respiratory tract (Broggi et al., 2020b; Major et al., 2020).
As mentioned earlier, the timing of IFNλ production may be a contributor to this difference—if it is relatively late in bacterial infections, as seen for B. pertussis infection, for example (Ardanuy et al., 2020), then adverse effects on neutrophil function and epithelial repair may be observed, whereas early production may limit inflammation and associated damage. In addition, it is possible that IFNλ signaling pathways are different in intestinal versus respiratory epithelial cells, which could be a function of the density of IFNλ receptors, which is apparently far higher in intestinal cells than in respiratory cells (Linden et al., 2020). This may explain the disparate observations of IFNλ-mediated decrease in tight junction protein expression in the respiratory epithelium but increase in expression in the intestinal tract.
However, this dichotomy may not be valid in all cases, since Broquet et al. (2020) found that administration of IFNλ to mice during P. aeruginosa pneumonia improves outcomes by inhibiting neutrophil recruitment and reducing epithelial damage, contrasting effects to those seen by others (as described earlier). This mix of apparently contrasting findings may be explained by context-specific pathogen and host factors that influence IFNλ signaling outcomes. Clearly, further investigation of downstream effects of IFNλ signaling and the activities of ISG products will be needed to resolve this issue.
Bacterial Suppression of IFNλ Responses
A well-studied phenomenon is the IFN suppressing and blocking activities of viruses to promote their infection (Hoffmann et al., 2015). Recent studies have demonstrated that the same may be true for certain bacterial infections. Sorenson et al. found that P. aeruginosa could reduce the level of IFNλ protein in the supernatant of virus-infected cultured cells and their results implicated the quorum sensing-dependent secreted protease AprA in this process, presumably by degrading IFNλ protein (Peignier and Parker, 2020; Sorensen et al., 2020).
It is not clear how this specific activity might benefit the bacterial pathogen, since P. aeruginosa grows to higher bacterial loads in the presence of IFNλ signaling than in its absence (Cohen and Prince, 2013), but these authors found that the virus infection status of cystic fibrosis patients correlated with the IFNλ-degrading activity of their P. aeruginosa isolates, demonstrating another important interplay between bacterial and viral infections in IFNλ-dependent outcomes. In another study, Alphonse et al. (2022b) found that although IFNλ restricts Shigella infection in intestinal epithelial cells, the bacteria can inhibit IFNλ signaling responses through the OspC effectors secreted into host cells by its type III secretion system.
The OspC proteins bound the calcium sensor calmodulin, which blocked calmodulin kinase II activity and downstream JAK/STAT signaling, thus suppressing IFNλ responses. OspC mutants of Shigella were attenuated in cultured intestinal epithelial cells and in mice, and this defect was rescued by IFNλ receptor deficiency, demonstrating the importance of targeting IFNλ signaling for the Shigella OspC proteins. They also observed that OspC homologs are present in other bacterial pathogens and had IFNλ inhibitory activity. Therefore, suppressive activity against IFNλ responses may be a more common property of bacterial pathogens, particularly for enteric pathogens where IFNλ may play a protective role. It will be interesting to determine whether respiratory pathogens, for which IFNλ promotes their growth and pathogenesis, have mechanisms to enhance IFNλ signaling or its effects, in contrast to the IFNλ-suppressing activities described in this study.
Conclusions
Our understanding of the roles of IFNλ signaling in bacterial infections is still in the early stages of investigation. Much basic information, such as the cell types that produce and respond to IFNλ, the specific ISGs that mediate activities, and the downstream effects that impact the infection, remains to be determined. However, certain themes appear to be emerging in this field. One is the effects of IFNλ on epithelial barrier integrity and the dichotomy of outcomes in respiratory versus gastrointestinal bacterial pathogens (Fig. 1).
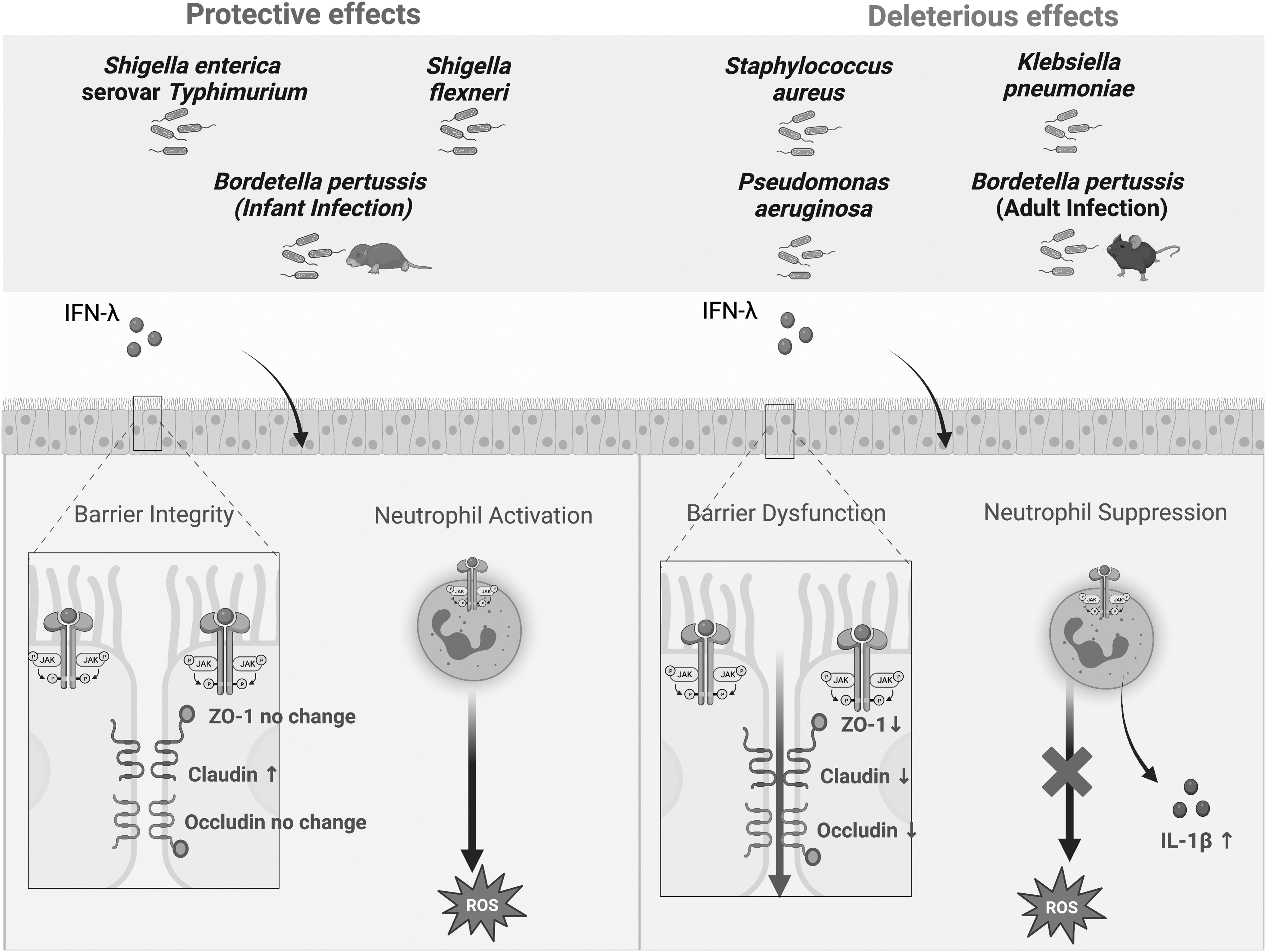
IFNλ signaling can be protective or deleterious depending on the pathogen. The bacterial infection models that showcase protective effects occur at the gastrointestinal epithelium, except for infant Bordetella pertussis infections. Deleterious effects occur at the respiratory tract. IFNλ directly binds and signals through its heterodimeric receptor on epithelial cells and neutrophils, resulting in changes to barrier integrity and neutrophil activation, respectively. Created with
Further research will be needed on specific mechanisms that result in changes to tight junction protein expression and how this differs between different epithelial compartments. Another theme is the effect of IFNλ on neutrophil activities, where research will be focused on signaling pathways and ISG effects that result in disparate outcomes for different microbial pathogens. The larger literature on the role of IFNλ in viral infections can be a useful guide, but there are clearly differences in IFNλ effects between viral and bacterial infections and the complexity is even higher when considering mixed infections or bacterial superinfection after influenza.
Yet another issue is age dependence of outcomes from IFNλ signaling. For instance, a recent study on the role of IFNλ signaling in mice infected with B. pertussis revealed distinct age-dependent outcomes. IFNλ expression was upregulated in the lungs of infected adult mice and exacerbated pulmonary inflammation, whereas infected infant mice (7 days old at inoculation) did not upregulate IFNλ expression and succumbed to the infection (Ardanuy et al., 2020). In contrast to the deleterious effects of IFNλ signaling in B. pertussis-infected adult mice, it is possible that IFNλ would be protective for infants against this infection (Fig. 1).
Treatment of infant mice with IFNλ protects against various viral infections (Lin et al., 2016; Mahlakõiv et al., 2015; Werder et al., 2018), indicating functionality of the IFNλ receptor in infants. The lack of IFNλ induction in infants may reflect the generally poor inflammatory responses to microbial ligands that increase with age (Lissner et al., 2015; Wang et al., 2017), but the specific point at which IFNλ responses are blocked is unclear. This raises another interesting point, in that this would represent a beneficial effect of IFNλ in the respiratory tract, in contrast to the deleterious effects seen in other bacterial respiratory infections described earlier. The age dependency of this transition from beneficial to deleterious effects may be related to the development of the epithelial tissues with increasing age after birth.
An important question will be whether IFNλ signaling represents a possible therapeutic target for some bacterial infections, as is being tested for certain viral infections, including COVID (Feld et al., 2021). This will depend on whether IFNλ effects are deleterious or beneficial for the particular infection and considerations of unintended effects on other pathogens, particularly viruses, will have to be included. In addition, the age of the patient may be a factor, since a beneficial treatment for an infant may be deleterious for an adult, as indicated from the B. pertussis infection model. It is our hope that this review will spur further investigation of the biology of IFNλ in the context of bacterial infections and considerations of its therapeutic potential.
Footnotes
Acknowledgments
The authors thank Carbonetti lab members for helpful discussions and Ashley Mitchell for help with the figure design.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by NIH grants AI141372, AI141372-S1, and AI168603.