
Abstract
Type III interferons (IFN-λ) are central to host defense against viral infection of epithelial barrier surfaces. IFN-λ binding to its receptor induces a JAK-STAT cascade through kinases Janus-associated kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), which are associated on either subunit of the heterodimeric type III IFN receptor. Recent studies have shown that TYK2 is not necessary for IFN-λ to signal, in contrast to IFN-α, which uses the same JAK-STAT pathway activated by the type I IFN receptor. The mechanism for this differential TYK2 requirement is unknown. Our study uses synthetic IFN receptors in TYK2-deficient U2OS epithelial cells to define the processes in type I and III IFN signaling that require TYK2. We find that TYK2 deficiency reduces signaling equally from heterodimers of either type I or III IFN receptor intracellular domains. In contrast, JAK1-associated homodimers of IFNAR2 or IFNLR1 are both fully signaling competent even in the absence of TYK2. These results suggest that heterodimerization of the type III IFN receptor is insufficient to confer TYK2-independent signaling. Thus, we propose that noncanonical receptor complexes may participate in endogenous type III IFN signaling to confer TYK2-independent signaling downstream of IFN-λ stimulation.
Introduction
Type III interferons (IFN-λs) play a vital role in the innate immune response to viral infection (Donnelly and Kotenko, 2010). Given that their receptor is expressed predominately on cells of epithelial origin, IFN-λ signaling occurs primarily at barrier surfaces, including the respiratory tract, gastrointestinal tract, liver, skin, placenta, and blood–brain barrier (Lazear et al., 2015b; Sommereyns et al., 2008; Wells and Coyne, 2018).
As such, IFN-λs induce potent antiviral responses against viruses such as hepatitis C virus (HCV), hepatitis B virus (HBV), yellow fever virus, West Nile virus, influenza virus, and SARS-CoV-2 (Anggakusuma et al., 2015; Douam et al., 2017; Felgenhauer et al., 2020; Galani et al., 2017; Klinkhammer et al., 2018; Lazear et al., 2015a; Marcello et al., 2006; Robek et al., 2005). This effective yet restricted antiviral signaling positions IFN-λ as a potential therapeutic alternative to type I IFNs against a range of infections, such as HCV, HBV, and SARS-CoV-2 (Flisiak et al., 2016; Phillips et al., 2017; Sohn et al., 2021).
Antiviral action of IFN-λ is mediated through a JAK-STAT signaling pathway that begins with activation of Janus-associated kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), each associated with the intracellular domains of the type III IFN receptor (O'Shea et al., 2013a; Philips et al., 2022).
Specifically, it has been shown that JAK1 associates with the IFNLR1 subunit (Dumoutier et al., 2004; Zhang et al., 2016), while TYK2 associates with IL10RB (Kotenko et al., 1997; Kotenko et al., 1996), a subunit also shared with the IL10 receptor. These molecular events parallel those that occur in type I IFN receptors, where JAK1 associates with IFNAR2 and TYK2 associates with IFNAR1. For both type I and III receptors, activation of JAK1 and TYK2 leads to subsequent phosphorylation of signal transducer and activator of transcription (STAT) proteins, which translocate into the nucleus and induce transcription of IFN-stimulated genes (ISGs) that regulate viral clearance.
Given their widespread role in cytokine signaling, JAKs are becoming a common therapeutic target for immune-mediated diseases (Damsky et al., 2021; O'Shea et al., 2013b). While many therapies predominately target JAK1, JAK2, and JAK3 (Lin et al., 2020), there has been recent interest in specifically targeting TYK2 to treat autoimmune disorders (Catlett et al., 2022; Catlett et al., 2019; Mease et al., 2022). Although this kinase presents as an attractive drug target, much is still unknown about how TYK2 inhibition or loss affects overall immune signaling in the context of infection. As such, it is crucial that more work be done to characterize its role in cytokine and IFN signaling.
TYK2 deficiency in patients is a rare disorder that correlates with increased susceptibility to certain viral and bacterial infections, as well as decreased IFN-α/β signaling (Meyts and Casanova, 2021; Minegishi et al., 2006); however, loss of TYK2 does not seem to affect IFN-λ signaling (Kreins et al., 2015). Studies in TYK2-deficient cell lines have confirmed that IFN-λs can induce STAT1 phosphorylation, ISG expression, and viral clearance in the absence of TYK2 (Fuchs et al., 2016; Schnepf et al., 2021).
Likewise, IFN-λ signaling can occur in the presence of a chemical inhibitor of TYK2 but not JAK1 or JAK2 (Schnepf et al., 2021). This has raised the question: Why can the type III IFN receptor signal without TYK2, particularly given their close molecular correspondence to type I IFN receptors whose signaling is reduced under the same conditions?
Here, we combined synthetic type I and III IFN receptor variants with CRISPR knockout of TYK2 to test how type III interferon receptors might support TYK2-independent signaling. We recently developed chimeric variants of type I and III IFN receptors that exclusively form homodimers or heterodimers in response to an orthogonal ligand, erythropoietin (EPO) (Mesev et al., 2023). Here, we engineered wild-type or TYK2-deficient cell lines expressing these receptors to characterize their signaling responses.
We found that loss of TYK2 dramatically reduced STAT1 phosphorylation downstream of forced heterodimers of either type I (IFNAR1 and IFNAR2) or type III (IL10RB and IFNLR1) intracellular domains. In contrast, synthetic homodimers of IFNAR2 or IFNLR1, as well as endogenous type III IFN receptors, each elicited a robust STAT response in TYK2-deficient cells.
Given that synthetic type III heterodimers cannot signal in the absence of TYK2 like their endogenous counterparts or IFNLR1 homodimers, our work raises the possibility that endogenous IFN-λ signaling may occur through noncanonical complexes of type III IFN receptor subunits. These may include multimeric clusters of IFNLR1/IL10RB or heterodimers that involve subunits other than IL10RB, suggesting additional routes through which IFN-λ signaling differs from IFN-α/β.
Materials and Methods
Reagents and cell cultures
All cells (U2OS, 293T-lentiX) were grown under standard conditions [37°C, 5% (v/v) carbon dioxide (CO2)] and cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% (v/v) heat-inactivated fetal bovine serum and 1% v/v penicillin/streptomycin (Corning Inc., NY, USA). Recombinant cytokines were purchased from Sigma-Aldrich [hEPO (E5546); hIFN-β (IF014); and hIFN-α2A (SRP4594); St. Louis, MO, USA] or R&D Systems [hIFN-λ1 (1598-1L); hIFN-λ2 (1587-IL); and hIFN-λ3 (5259-IL); Minneapolis, MN, USA].
Plasmid construction
Plasmids for human STAT1 (No. 12301; Timofeeva et al., 2006) and pDONR223-TYK2 (No. 23908; Johannessen et al., 2010) were purchased from Addgene (Watertown, MA, USA). All plasmid backbones were linearized using restriction enzymes or polymerase chain reaction (PCR) amplification, and inserts were cloned by PCR amplification. Assembly reactions were performed using the inFusion kit (Clontech, Mountain View, CA, USA), and stellar competent Escherichia coli cells (Clontech) were transformed according to manufacturer's instructions. All final plasmids were validated by sequencing (Genewiz, South Plainfield, NJ, USA).
TYK2 CRISPR
Forward TYK2 sgRNA oligo: CACCGAACCGGCTGTGTACCGTTG
Reverse TYK2 sgRNA oligo: AAACCAACGGTACACAGCCGGTTC
Annealed oligos were inserted into a BsmB1-digested V2 lenti CRISPR backbone encoding Cas9 and puromycin resistance gene. The final plasmid was inserted into U2OS cells using lentiviral transduction, and 72 h later cells were cultured in 1 μg/mL puromycin for 14 days to select for transduced cells only. A bulk population of puromycin-resistant cells were then validated for experiments in this study: targeting of the TYK2 locus was determined by genomic DNA sequencing, and TYK2 protein expression was analyzed by western blot.
Lentivirus production and transduction
HEK293T-lentiX cells were grown to 90% confluency on poly-L-lysine-coated plates, then cotransfected with plasmid of interest (0.67 μg) and lentiviral packaging plasmids VSV-G (0.095 μg) and HIV Gal-Pol (0.67 μg) using X-tremeGENE9 transfection reagent (Sigma-Aldrich). Supernatants were collected after 48 h, filtered through 0.45 μm filter, and supplemented with 1:1000 polybrene (stock 4 mg/mL) and 1:50 HEPES (stock 1 M) before transduction of cells for 18–24 h. Experiments were performed >48 h later to ensure stable plasmid integration.
Western blotting
Cells were lysed in RPPA buffer [1% Triton X-100, 50 mM HEPES buffer, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM sodium pyrophosphate, 1 mM Na3VO4, 10% (v/v) glycerol] supplemented with protease and phosphatase inhibitors, before being mixed with 6 × Laemmli buffer/2-mercaptoethanol, heated at 95°C for 5 min, and loaded into a 4%–12% Bis-Tris gel (Invitrogen, Waltham, MA, USA) for electrophoresis.
Gels were transferred into a nitrocellulose membrane using the iBlot dry transfer system (Thermo Fisher Scientific), blocked in Tris-buffered saline with 0.1% (v/v) Tween 20 (TBST) with 5% (v/v) milk for 30 min at room temperature, and incubated overnight in antibodies specific for phospho-STAT1 (1:1,000; CST 9167), tSTAT1 (1:1,000; CST 9176 or CST 14994), or β-actin (1:2,500; CST 3700) purchased from Cell Signaling Technology (Danvers, MA, USA). Blots were then incubated for 1 h in either IRDye 680 CW or 800 CW secondary antibodies (Licor, Lincoln, NE, USA), and imaged with the Li-Cor Odyssey Infrared Imaging System (Licor), and signal intensities were quantified using ImageJ software.
Confocal microscopy
Cells were plated 24 h before imaging on collagen-coating 0.17 mm glass-bottomed, black-walled 96-well plates (In Vitro Scientific, Mountain View, CA, USA). Mineral oil was added before imaging to prevent evaporation, and cells were kept at 37°C with 5% (v/v) CO2 while being imaged. All imaging was performed using a Nikon Eclipse Ti microscope with a Prior linear motorized stage, a Yokogawa CSU-X1 spinning disk, an Agilent laser line module containing 405, 488, 561, and 650 nm lasers, and iXon DU897 EMCCD camera, and a 60 × oil immersion objective lens.
RT-quantitative real time PCR
RNA was extracted from cells using Monarch Total RNA Miniprep kit (Monarch Biosciences, Los Angeles, CA, USA). cDNA synthesis and RT-qPCR were performed using Luna Universal One-Step RT-qPCR Kit (NEB, Ipswich, MA). Cycle threshold (Ct) values for genes of interest were normalized to HPRT1 as a control for input RNA.
Oligos for RT-qPCR
HPRT1 Forward Primer: ACTGAAGAGCTATTGTAATGACCAG
HPRT1 Reverse Primer: TGGATTATACTGCCTGACCAAG
MX1 Forward Primer: GTTTCCGAAGTGGACATCGCA
MX1 Reverse Primer: CTGCACAGGTTGTTCTCAGC
PKR Forward Primer: GCCGCTAAACTTGCATATCTTCA
PKR Reverse Primer: TCACACGTAGTAGCAAAAGAACC
Statistical analysis
Statistical analyses were performed with GraphPad Prism 8 software and presented as mean ± standard error. Significance between groups was determined by one-way (where only one cell type was used) or two-way (where multiple cell types and treatment conditions were used) analysis of variance with the Tukey–Kramer post-test (multiple comparisons), and difference between means was considered statistically significant at P < 0.05.
Results
Signaling by IFN-λ subtypes is unaffected in Tyk2–/– U2OS epithelial cells
Prior studies have suggested that type I and III IFN receptors are differentially susceptible to TYK2 loss (Fuchs et al., 2016; Kreins et al., 2015; Schnepf et al., 2021). We first set out to confirm this differential requirement by constructing a Tyk2–/– variant using CRISPR-Cas9 in U2OS human epithelial cells, which express both type I and III IFN receptors, and therefore can be used to compare signaling by type I and III IFNs. We used a lentivirally expressed CRISPR-Cas9 system to generate Tyk2–/– U2OS cells with an sgRNA targeting Exon 3 of the Tyk2 genes (Fig. 1A).
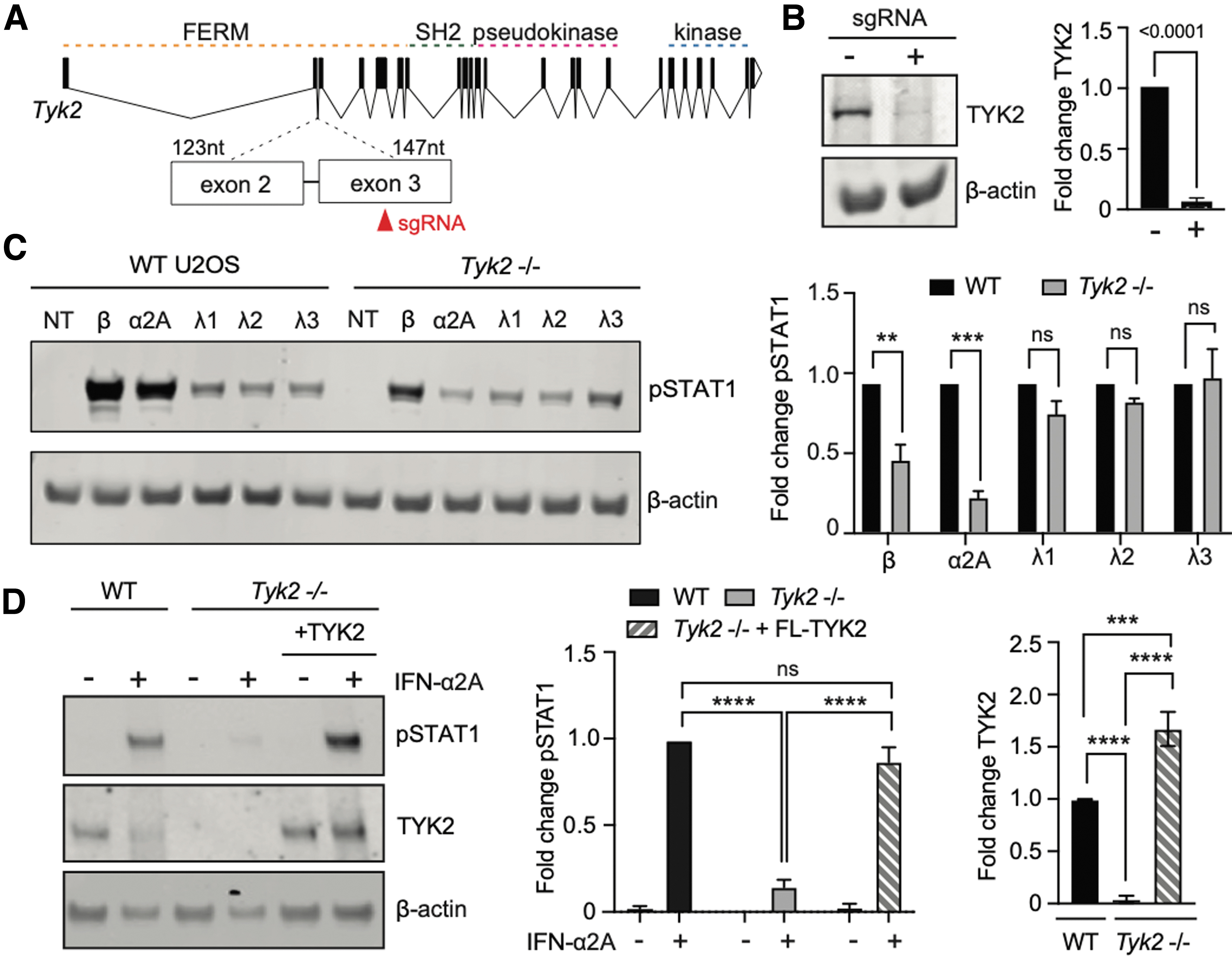
IFN-λ induces STAT1 phosphorylation regardless of TYK2 expression.
We bulk-enriched sgRNA-expressing cells using puromycin selection and tested for loss of TYK2 protein by Western blot, revealing successful loss of this protein product (Fig. 1B). These cells were used as a U2OS Tyk2–/– cell line for all subsequent experiments.
We next tested for the role of TYK2 in transducing type I and III IFN signaling in U2OS cells. We first determined saturating concentrations of IFNs that would activate maximal STAT1 phosphorylation through endogenous type I and III IFN receptors in U2OS cells (Supplementary Fig. S1A, B). We then treated wild-type or Tyk2–/– U2OS with saturating concentrations of various IFN subtypes (type I: IFN-α2A and IFN-β; type III: IFN-λ1, IFN-λ2, and IFN-λ3) and measured STAT1 phosphorylation (pSTAT1) after 30 min (Fig. 1C). We found that type I IFN stimulation drove higher levels of pSTAT1 than type III IFNs, consistent with prior reports (Fig. 1C, left).
In contrast to their wild-type counterparts, U2OS Tyk2–/– cells exhibited substantially diminished pSTAT1 in response to IFN-α2a and IFN-β treatment, but saturating concentrations of all three IFN-λ subtypes were still able to induce pSTAT1 to the same magnitude (Fig. 1C, right). We confirmed that our results were in fact due to TYK2 deficiency by transducing full-length (FL) TYK2 into Tyk2–/– cells. Indeed, rescuing TYK2 expression restored IFN-α signaling (Fig. 1D).
Altogether, these data confirm that TYK2 deficiency in U2OS cells substantially reduces IFN-α/β signaling but does not affect STAT1 phosphorylation by IFN-λ1, λ2, or λ3 at saturating concentrations. Of note, supersaturating concentrations of IFN-α2A induced slightly higher levels of pSTAT1 in Tyk2–/– cells, despite having maximized the levels of pSTAT1 in wild-type U2OS cells (Supplementary Fig. S1C).
Interestingly, subsaturating concentrations of IFN-λ3 led to reduced pSTAT1 levels in Tyk2–/– cells, even though saturating doses showed similar responses regardless of TYK2 status (Supplementary Fig. S1C). These results suggest that the mechanism responsible for TYK2-independent signaling may rely on activation of higher numbers of the type III IFN receptors on the cell surface.
Heterodimeric type III IFN receptor chimeras require TYK2 for STAT1 activation
Since TYK2 is known to associate with the IL10RB subunit of the type III IFN receptor complex, we hypothesized that type III IFN receptors may support TYK2-independent signaling through one of two mechanisms: (i) IL10RB may associate with a different JAK kinase in the absence of TYK2, or (ii) IFNLR1 may form noncanonical complexes or clusters that allow it to signal even when IL10RB is not bound to any kinase.
To begin testing these hypotheses, we utilized synthetic, heterodimeric receptors that we previously engineered to characterize the signaling capabilities of type I and III IFN receptors (Mesev et al., 2023; Fig. 2A). In these receptors, the intracellular and transmembrane domains of both IFN receptors are fused to variants of the extracellular domain of the EPO receptor (EPOR).
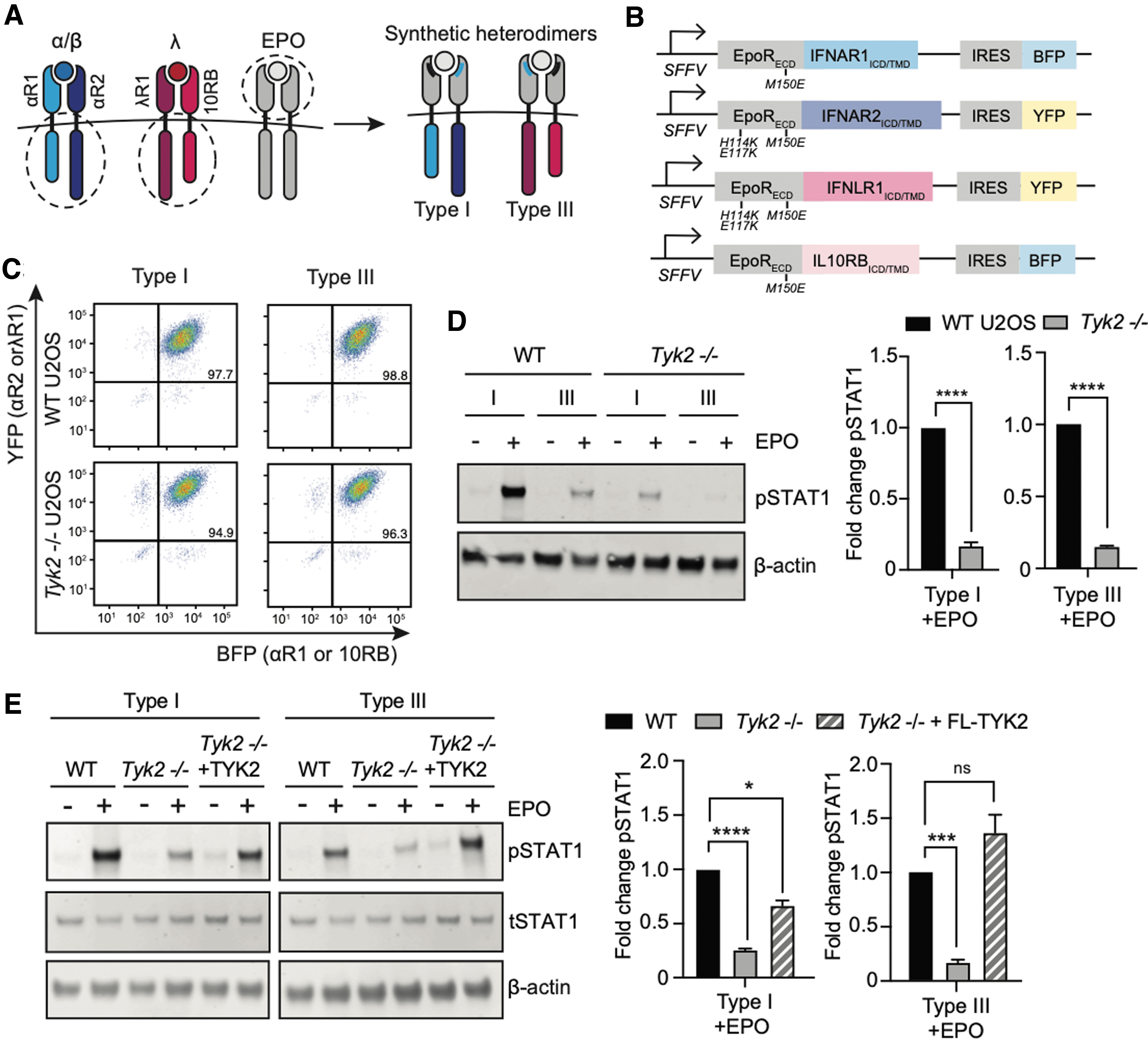
Synthetic heterodimers of type I and III IFN receptors exhibit impaired STAT1 phosphorylation.
The natural EPOR can bind its ligand EPO at two distinct sites, forming an asymmetric homodimer complex (Zhang et al., 2009). Here, we mutated each EPOR-IFN receptor fusion protein to only a single site on the EPO ligand, thus creating an obligate heterodimeric complex upon signaling stimulation. We predicted that forcing heterodimers of IFNLR1-IL10RB in TYK2-deficient cells would permit signaling only if another kinase could compensate for the lack of TYK2 on IL10RB.
We stably transduced wild-type and Tyk2–/– U2OS cells with either our synthetic type I or III heterodimeric IFN receptor systems. Each subunit expressed either a yellow fluorescent protein (YFP) or blue fluorescent protein (BFP) downstream of an internal ribosome entry site (IRES) (Fig. 2B), which allowed us to sort for similar total receptor expression levels using flow cytometry (Fig. 2C). The resulting cell lines expressed either a type I or III IFN receptor pair whose heterodimerization can be induced by EPO ligand stimulation, in either a wild-type or Tyk2–/– background.
To characterize their signaling, we treated each cell line with 100 ng/mL EPO for 30 min. Just as in the case of endogenous IFN receptor stimulation (Fig. 1C), we observed a ∼5-fold difference in pSTAT1 between our synthetic type I and III IFN receptors after EPO stimulation (Fig. 2D, left). This result is also consistent with our previous observations using synthetic type I and III IFN receptors (Mesev et al., 2023).
Unlike the endogenous type III IFN receptors, however, our synthetic heterodimeric receptors exhibited dramatically reduced STAT1 phosphorylation on a Tyk2–/– background (Fig. 2D). When normalized to wild-type levels, this reduction was the same as that observed for the synthetic type I IFN receptor, indicating that both type I and III heterodimeric IFN receptors were equally impaired by the loss of TYK2 (Fig. 2D).
We again confirmed that this signaling impairment was specifically caused by TYK2 loss by reintroducing FL-TYK2 into the Tyk2–/– cell lines expressing synthetic type I and III IFN receptors and repeating our EPO stimulation experiment (Fig. 2E). Indeed, expression of exogenous TYK2 rescued signaling by both synthetic type I and III heterodimeric IFN receptors, confirming that loss of TYK2 is sufficient to ablate STAT1 phosphorylation by heterodimeric type I and III IFN receptors.
Noncanonical IFN receptor pairs reveal TYK2-independent signaling complexes
The canonical model of IFN signaling holds that TYK2 associates with homologous chains of both type I and III receptors: IFNAR1 and IL10RB respectively. To further test whether both chains were equally susceptible to TYK2 loss, we carried out a receptor swap experiment by driving heterodimerization between one type I and one type III IFN receptor subunit.
We thus generated cell lines expressing synthetic heterodimerizing pairs of IFNAR1 and IFNLR1 subunits, or IL10RB and IFNAR2 subunits, in either wild-type or Tyk2–/– cells (Fig. 3A). As done previously for our synthetic type I and III IFN receptor heterodimers, cells were sorted for similar expression levels before being treated with EPO (100 ng/mL) for 30 min (Fig. 3B).
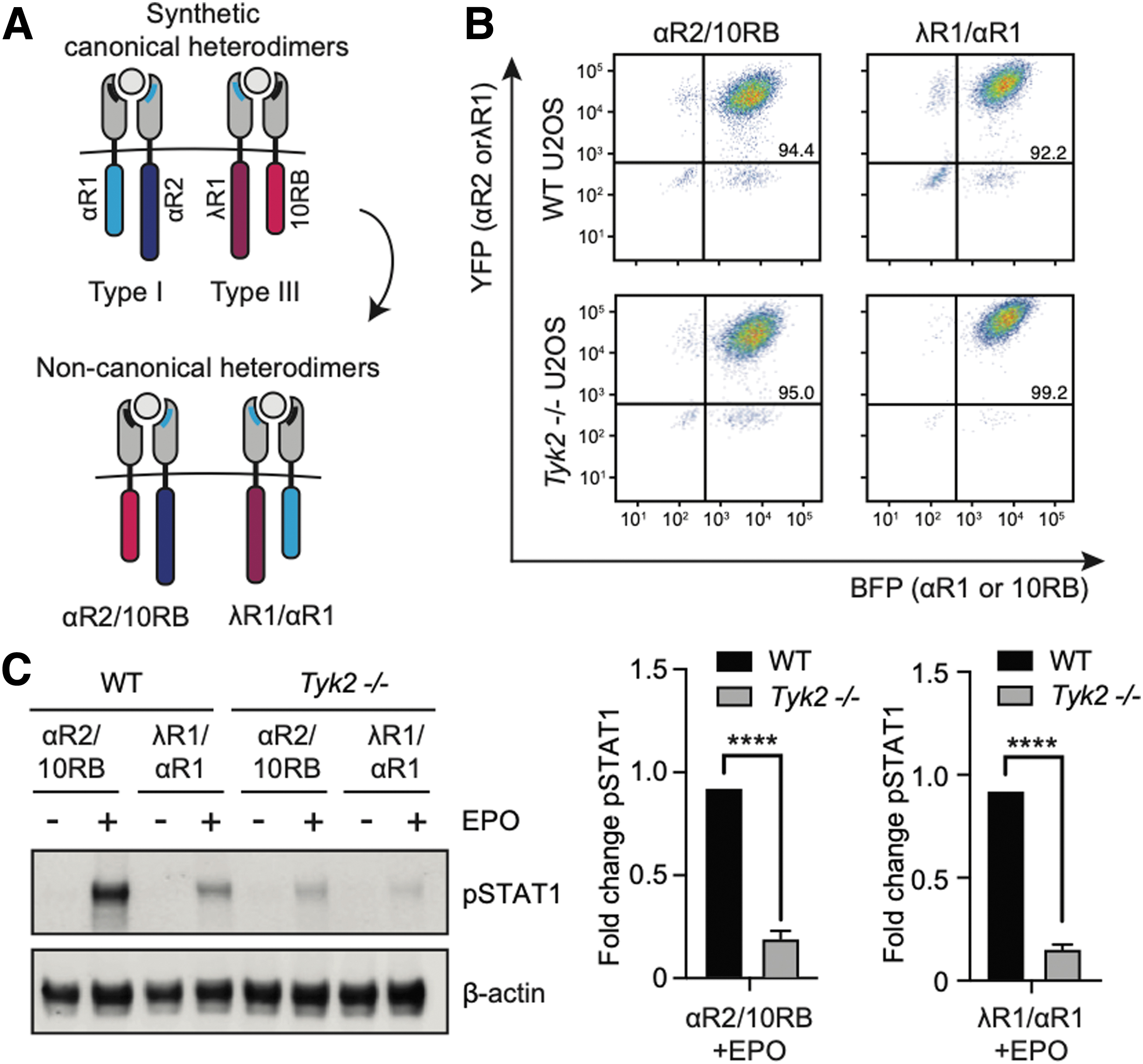
Noncanonical receptor pairings of synthetic IFN receptor subunits do not permit STAT1 phosphorylation in TYK2-deficient cells.
Just as in the case of synthetic type I and III IFN receptor heterodimers, we found that these type I/III mixed heterodimers were each equally sensitive to TYK2 loss (Fig. 3C). These results are broadly consistent with a model where IL10RB and IFNAR1 are similarly dependent on TYK2 association, as exchanging these subunits has a negligible effect on signaling outcome. These results are unexpected: whereas endogenous type III signaling is robust to TYK2 deficiency, individual type III IFN receptor subunits are not.
The preceding experiments raise a conundrum: if no heterodimeric IFN receptor complexes can signal potently in the absence of TYK2, what receptor complexes might be formed in the endogenous context to confer a potent type III response in Tyk2–/– cells? We hypothesize this capability might be conferred by noncanonical complexes containing two or more IFNLR1 subunits, based on the observation that synthetic IFNAR2 or IFNLR1 homodimers, neither of which engage with TYK2, are capable of potent signaling (Mesev et al., 2023).
To test this hypothesis, we generated chimeric fusions of the IFNAR2 and IFNLR1 intracellular domains with the unmutated extracellular domain of EPOR (Fig. 4A), which naturally assembles as a homodimer (Watowich et al., 1992). Previous studies by various groups, including ours, have constructed similar EPOR chimeras to characterize homodimeric signaling of IFN receptor subunits (Muthukumaran et al., 1997; Pattyn et al., 1999).

IFNLR1 and IFNAR2 homodimer signaling is unaffected by TYK2 expression.
We transduced our EPOR-IFNAR2 or EPOR-IFNLR1 fusions into wild-type or Tyk2–/– U2OS cells and sorted for similar expression levels by flow cytometry using an IRES-YFP tag on either construct (Fig. 4B). We again stimulated cells with EPO for 30 min before assessing STAT1 phosphorylation levels.
Unlike our prior synthetic heterodimers, we found that synthetic EPOR-IFNAR2 and EPOR-IFNLR1 homodimers were both able to induce robust STAT1 phosphorylation in Tyk2–/– U2OS cells, exhibiting indistinguishable responses to their TYK2-containing counterparts. (Fig. 4C, D). IFNAR2 and IFNLR1 homodimers also led to robust STAT2 phosphorylation (Fig. 4E).
Furthermore, when analyzed on the same western blot, homodimers were found to have a stronger pSTAT1 response than synthetic type I and III IFN receptor heterodimers (Fig. 4E). In sum, our data demonstrate that although canonical heterodimers of either type I or III IFN receptors are equally susceptible to TYK2 loss, homodimers of either IFNAR2 or IFNLR1 intracellular domains are capable of TYK2-independent signaling, suggesting a potential route by which endogenous IFN receptors might circumvent the requirement for TYK2.
Type I and III IFN receptor homodimers elicit potent JAK-STAT signal transduction
We next set out to characterize the dynamic range with which type I and III homodimeric subunits signal and to measure their responses over time. We first performed a dose–response curve in Tyk2–/– cells expressing EPOR-IFNAR2 and EPOR-IFNLR1 by treating cells with EPO concentrations spanning two orders of magnitude (0.5–50 ng/mL). Although signaling by the EPOR-IFNAR2 homodimer was always more potent than the EPOR-IFNLR1 homodimer, both homodimers were able to induce strong levels of STAT1 phosphorylation in Tyk2–/– cells, especially at higher concentrations of EPO (Fig. 5A).

IFNAR2 and IFNLR1 induce robust STAT1 activation and translocation in the absence of TYK2.
We performed a similar dose–response experiment with type I and III heterodimers in wild-type cells and observed a 3-fold difference in pSTAT1 across all EPO doses tested (Fig. 5B). These data suggest that pSTAT1 levels induced by IFN receptor homodimers are more similar than pSTAT1 levels induced by heterodimeric type I and III IFN receptors. More broadly, we posit that the intracellular domains of IFNAR2 and IFNLR1 are each capable of inducing potent STAT1 responses on their own, upon proper dimerization, even in the absence of a heterodimerizing binding partner.
To further characterize signaling dynamics upon EPOR-IFNAR2 and EPOR-IFNLR1 homodimerization, we took advantage of an mCherry-STAT1 fluorescent reporter that can be visualized in cells (Köster and Hauser, 1999). STAT1 translocates from the cytoplasm to the nucleus upon phosphorylation by IFN receptors, as essential step for the propagation of IFN signaling to gene expression (Meyer and Vinkemeier, 2004). We transduced our Tyk2–/– U2OS cells with an mCherry-STAT1 construct that allowed us to visualize STAT1 translocation into the nucleus by confocal microscopy upon EPO stimulation (Fig. 5C).
Although the lowest concentrations of EPO produced weaker STAT1 translocation in cells expressing EPOR-IFNLR1 homodimers, higher concentrations of EPO allowed both homodimers to induce similar levels and dynamics of nuclear STAT1 accumulation when normalized to their respective starting levels (Fig. 5D). Nuclear translocation of STAT1 plateaued at 2–4 h after EPO stimulation for both homodimers (Fig. 5D). Altogether, these results suggest that maximal induction of IFNAR2 and IFNLR1 homodimers is sufficient to activate robust STAT1 nuclear translocation—an important step in JAK-STAT signal transduction after STAT phosphorylation.
Finally, we performed RT-qPCR to assess whether IFNAR2 and IFNLR1 homodimers were able to induce transcription of ISGs. Tyk2–/– U2OS cells stably expressing EPOR-IFNAR2 or EPOR-IFNLR1 were treated with EPO (100 ng/mL) for 8 h. For comparison, we also treated wild-type or Tyk2–/– U2OS cells with IFN-α2A (1000 IU/mL) in parallel for 8 h. We then extracted RNA from the cell lysates and performed RT-qPCR to measure relative levels of ISGs.
As expected, Tyk2–/– cells stimulated with IFN-a2A led to a nonsignificant change in ISG induction, while activation of IFNAR2 and IFNLR1 homodimers led to 6-fold induction of PKR and 4-fold induction of MX1 compared with untreated cells (Fig. 5E). Overall, these data demonstrate that homodimeric complexes of either IFNAR2 or IFNLR1 are indeed capable of robust signaling and gene expression through STAT even in the absence of TYK2. It is therefore conceivable that these subunits may be able to signal if clusters of IFN receptors are indeed able to form endogenously.
Discussion
Our study aimed to address the question of why IFN-λ signaling unaffected in the absence of TYK2 (Fig. 6A). Using synthetic IFN receptors, we have shown that heterodimers of IFNLR1 and IL10RB are not sufficient for normal STAT activation in TYK2-deficient cells (Fig. 6B), nor is IL10RB able to “rescue” STAT activation when paired with IFNAR2 (Fig. 6C).
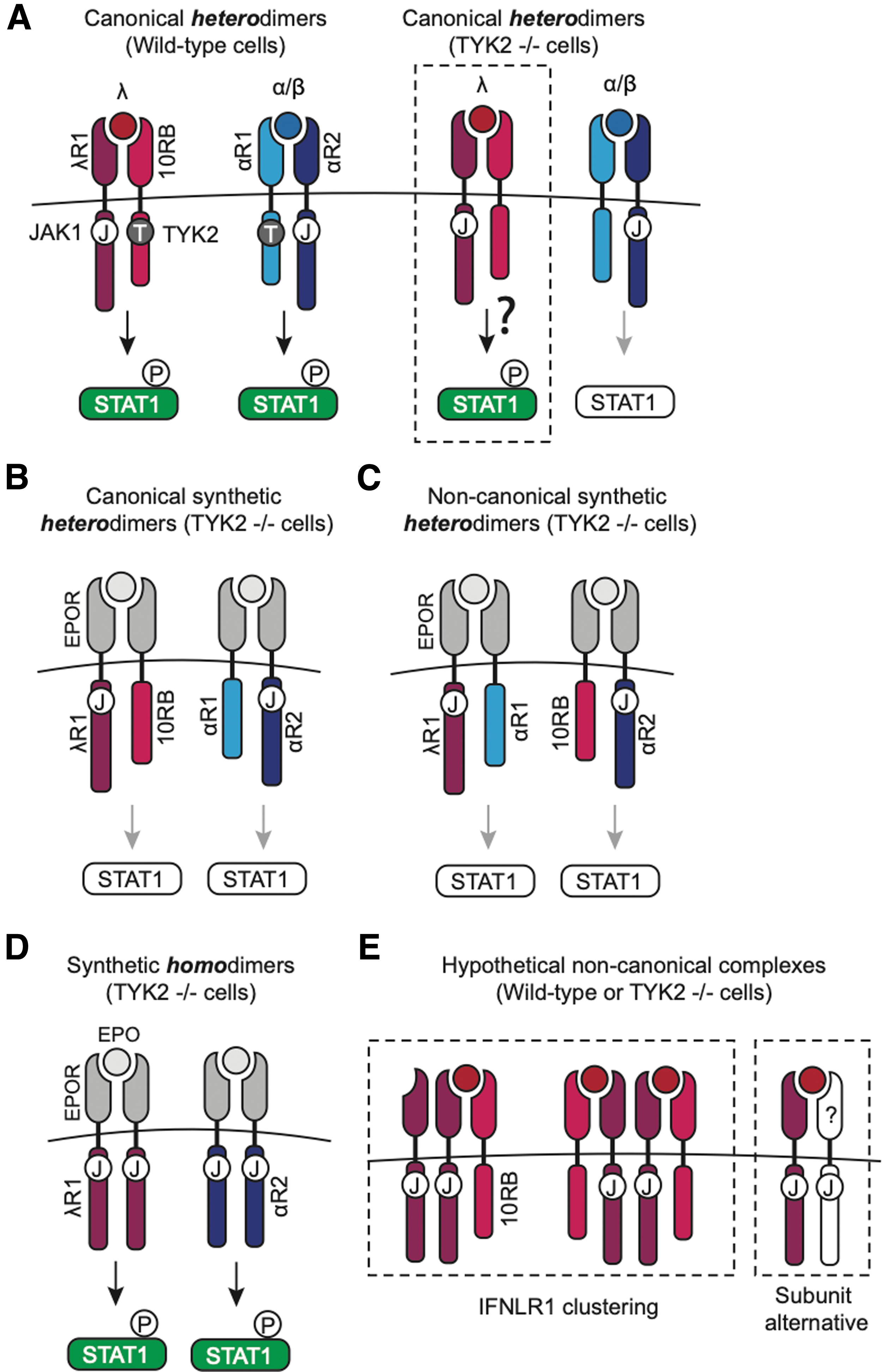
Models of IFN receptor signaling in the presence or absence of TYK2.
In contrast, IFNLR1 and IFNAR2 homodimers are able to induce robust STAT phosphorylation and translocation in wild-type and Tyk2–/– cells (Fig. 6D). Altogether, these data suggest that TYK2-independent IFN-λ signaling relies on the IFNLR1 subunit, which may be capable of forming noncanonical receptor assemblies that can signal in the absence of TYK2 and/or IL10RB.
While previous data suggest that IFNLR1 cannot form signaling-competent homodimers endogenously (Kotenko et al., 2003), we propose that alternative noncanonical complexes could exist beyond the standard heterodimer model, such as (a) clusters/oligomers of heterodimers (e.g., IFNLR1 bound to IL10RB); or (b) dimers of IFNLR1 with an alternative binding partner (Fig. 6E). Receptor clustering has been proposed for other cytokine receptors, such as the IL10 receptor, which appears capable of forming a 2:4 complex of IL10B:IL10RA subunits (Josephson et al., 2001).
Moreover, our observation that a stronger TYK2-independent response is achieved at high type III IFN doses (Supplementary Fig. S1C) is consistent with a receptor clustering model, as these conditions would also be expected to produce an increased surface density of ligand-bound receptors, favoring their oligomerization. Given that our study suggests that proximity of multiple IFNLR1 subunits can induce signaling, future work should investigate whether clustering may be possible for the type III IFN receptors.
It has been shown that loss of TYK2 leads to reduced IL10RB expression at the membrane (Fuchs et al., 2016), leading some to speculate that TYK2 is involved in stabilizing IL10RB, similarly to how TYK2 controls IFNAR1 surface expression (Ragimbeau et al., 2003). Further studies should compare whether TYK2 deficiency affects IL10RB and IFNAR1 expression differently, which may explain signaling differences. If TYK2 deficiency reduces IFNAR1 and IL10RB to the same extent, it is conceivable that IFN-λs may signal through complexes that only require low levels of IL10RB and several copies of IFNLR1.
Alternatively, while it has been previously shown that expression of IFNLR1 alone is not sufficient for IFN-λ signaling (Kotenko et al., 2003), this does not preclude the possibility that IFN-λs could bind other receptor subunits beyond IL10RB. Such promiscuity may allow IFN-λ signaling to proceed unabated in the absence of TYK2. Either of these two models warrants further investigation.
While our study suggests alternative assemblies for the type III IFN receptor, our synthetic receptors are limited in their ability to directly test these models. A system that uses the IFNLR1 extracellular domain, rather than EPOR, could be used to explore whether endogenous IFN-λ can induce clusters of the IFNLR1 subunit and/or IFNLR-IL10RB dimers.
Furthermore, it remains to be seen whether other subunits can compensate for the lack of IL10RB. The IL10 receptor (composed of IL10RA and IL10RB) also appears to have a limited requirement for TYK2 (Wilbers et al., 2017), which raises the question of whether the IL10 and type III IFN receptors have more in common than a shared subunit.
Future studies will be needed to test which noncanonical receptor complexes may form endogenously to allow IFN-λ signaling regardless of whether TYK2 is present. Future studies should also focus on extending our findings to cell types other than the U2OS cells used in this study, and should explore whether the fourth IFN-λ subtype, IFN-λ4, is likewise unaffected by TYK2 deficiency.
Not only does our work propose a noncanonical assembly for the type III IFN receptor in a wild-type context, but it contributes to our understanding of innate immune responses that may occur in patients with TYK2 deficiency or in cases where TYK2 is targeted therapeutically. With renewed interest in both JAK inhibitors and pegylated IFN-λ therapies, it is essential that these nuances in IFN signaling continue to be explored.
In addition to unaffected type III IFN signaling, a degree of residual STAT1 phosphorylation may still occur in TYK2-deficient cells in response to type I IFNs, as we observed with our CRISPR U2OS cell line. This suggests that other kinases may bind weakly to the type I IFN receptor when TYK2 is not present. While several studies have already begun characterizing IFN-induced antiviral responses in TYK2-deficient cells (Fuchs et al., 2016; Kreins et al., 2015; Schnepf et al., 2021), further studies are needed to fully understand the innate immune profile that occurs when this kinase is absent or chemically inhibited.
Our findings also raise important implications for synthetic receptor development in general. As synthetic biology has risen in popularity over recent decades, synthetic receptors have become increasingly valuable tools to interrogate signaling pathways, including but not limited to cytokine and interferon signaling (Chang and Bonnet, 2020; Manhas et al., 2022). Engineered receptors even have therapeutic potential (Bell and Gottschalk, 2021; Scheller et al., 2019).
Care, however, must be taken when designing synthetic receptors to fit a canonical mode. As we have proposed here, some receptors may form complexes beyond their canonical architecture, a nuance that could be lost in a synthetic system. To gain a complete picture of a given receptor-mediated signaling pathway, several synthetic systems may need to be engineered, which capture all permutations of the assembled receptor. However, therein also lies the great advantage of synthetic receptors: by engineering many iterations of the same receptor in various genetic contexts, we can uncover aspects of signaling that might otherwise remain unknown.
Footnotes
Acknowledgments
The authors thank Sergei Kotenko (Rutgers New Jersey Medical School), Britt Adamson, and all members of the A.P. and J.E.T. laboratories for their discussions about this project. They also thank Jan Tavernier (Ghent University) for sharing EPOR-IFNAR1 and EPOR-IFNAR2 expression plasmids, as well as Christina DeCoste and Katherine Rittenbach at the Flow Cytometry Facility for their assistance.
Authors' Contributions
E.V.M. contributed to conceptualization, experimentation, data analysis, preparation of results, primary writing, and editing of the article. E.G.G. assisted with experimentation, data analysis, and preparation of results. A.P. and J.E.T. provided funding procurement, conception and design, critical analysis, supervision, interpretation and editing of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Financial support was received from NIH (Grant Nos. T32GM007388 to E.V.M., R01AI138797, R01AI107301, R01AI146917, R01AI153236, R01AI168048 to A.P., and DP2EB04247 to J.E.T.); Burroughs Wellcome Fund (to A.P.); American Cancer Society (Grant No. RSG-15-048-01-MPC to A.P.); Vallee Scholars (to J.E.T.); and NSF (Grant No. 1750663 to J.E.T.). The Princeton University Flow Cytometry Resource Facility is supported, in part, with funding from NCI-CCSG P30CA072720-5921.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.