
Abstract
Among cytokines, interferons (IFNs) have demonstrated antitumor effects through several mechanisms, making them attractive candidates for cancer immunotherapies. However, the application of interferons in cancer treatment comes with some obstacles, such as high toxicity owing to systemic exposure and a short half-life. The mQXL138A1 fusion protein is a murine interferon alpha-1 (IFN-α-1) fused to an antimurine Syndecan-1 (CD138) antibody designed with the aim of targeted delivery of IFN-α-1 to CD138-expressing tumor cells to enhance treatment efficacy and decrease toxicity. mQXL138A1 maintained its CD138 binding properties and exhibited IFN-dependent activity in vitro. mQXL138A1 exhibited antitumor efficacy across various syngeneic mouse models and minor levels of toxicity. Tumor growth inhibition was achieved through a combination of direct tumor cell killing and a shift in the immune activation profile of the tumor microenvironment. Although this study showed that tumor-targeted IFN-α-1 had broad antitumor properties across a range of solid tumor indications, toxicities associated with systemic activity remain an issue.
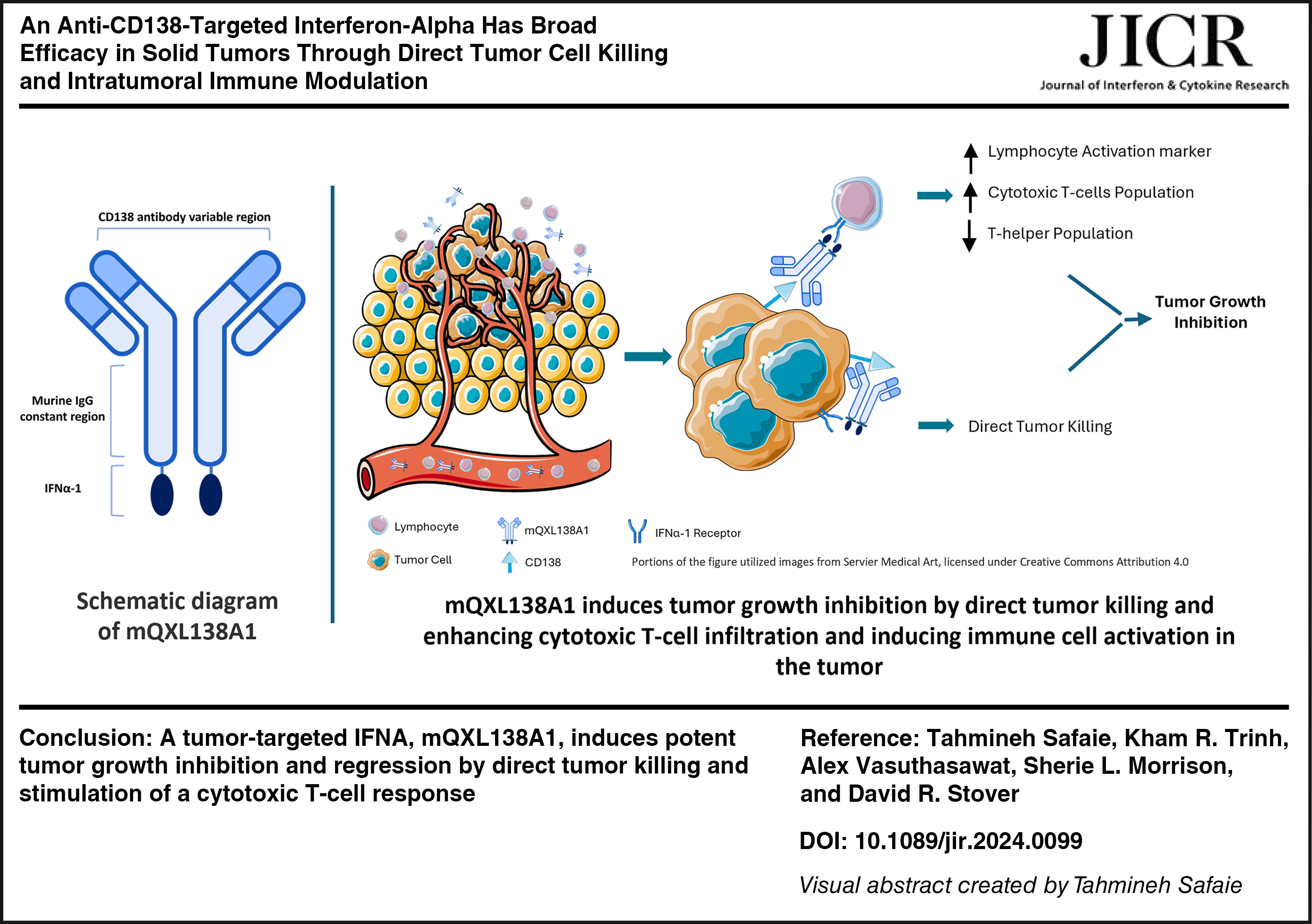
Introduction
Cytokines are powerful immune-modulating proteins that have been used in cancer treatment because of their antitumor activity and essential roles in the function and survival of immune cells (Holicek et al., 2023; Waldmann, 2018). Type I interferon (IFN) (specifically IFN-α-2) was the first cytokine approved for use in cancer treatment and has been used with some success in the treatment of several types of cancer, including hematological malignancies (multiple myeloma, hairy cell leukemia, chronic myeloid leukemia, and some B and T cell lymphomas) and solid tumors such as melanoma and renal carcinoma (Aricò et al., 2019; Fosså, 2000; Moschos and Kirkwood, 2007; Trinchieri, 2010). IFN-α exerts its antitumor effects through several mechanisms. In addition to direct inhibitory effects on cancer cell survival and proliferation, they enhance immune cell response to cancers through mechanisms such as increasing NK cell and T cell activity and tumor infiltration, increasing tumor antigen-specific CD8 cells in the peripheral blood, promoting the cross-activation of T cells and dendritic cells, reducing T-regulatory cell infiltration into tumors, decreasing differentiation and maturation of MDSCs, and blocking their suppressive activity on CD8+ T cells (Ferrantini et al., 2007; Xue et al., 2021; Yu et al., 2022).
Using IFNs in cancer treatment faces challenges, including systemic toxicity and a shortened duration of effectiveness owing to their short half-life. Many approaches have been explored to tackle these constraints, including the development of antibody–cytokine fusion proteins. These fusion proteins have been designed to leverage the tumor-targeting capabilities of monoclonal antibodies (mAbs) to precisely direct cytokines to tumor locations, potentially enhancing antitumor immune responses more effectively while mitigating the systemic toxicities that currently restrict cytokine dosing and, consequently, their potential effectiveness (Lechner et al., 2011; Ortiz-Sánchez et al., 2008; Young et al., 2014). IFNs have been conjugated to different mAbs that recognize different tumor-associated proteins and have been tested in vitro and in animal models. It was shown that the anti-HER2/neu antibody–IFN-α fusion protein had in vitro and in vivo efficacy against an experimental murine B cell lymphoma tumor overexpressing HER2/neu (Huang et al., 2007). In another study by Li et al, anti-VEGFR2-IFN-α-2 regulated the tumor microenvironment and showed potent antitumor efficacy against colorectal cancer (Li et al., 2017). Targeted delivery of IFN-α via fusion to anti-CD20 results in potent antitumor activity against B cell lymphomas (Trinh et al., 2013; Xuan et al., 2010). It has also been shown that IFN-α-14 fused to anti-CD138 antibody synergized with bortezomib to inhibit tumor growth in different multiple myeloma murine tumor models (Vasuthasawat et al., 2016).
We designed mQXL138A1 to target IFNs to cancer cells with the aim of selectively increasing antitumor potency while not simultaneously increasing toxicity. mQXL138A1 is a novel fusion protein composed of murine IFN-α-1 fused to a murine anti-Syndecan-1 (CD138) antibody. Although the approved therapies use human IFN-α-2, mice lack a clear homolog. We have used murine IFN-α-1 for this purpose as it is among the most conserved of the isotypes to human IFN-α-2 and has been reported to have antiproliferative and antiviral activity in the midrange for murine IFN-α isotypes (van Pesch et al., 2004). CD138 is a cell surface protein that is involved in cell proliferation, regulation, migration, and cytoskeletal organization (Kind et al., 2019). It has recently been found that IL-17-producing natural killer T cells (NKT17 cells) express CD138 (Liman and Park, 2023). Although CD138 is not expressed by most other normal lymphoid cells, it is expressed on various malignant hematological cells, including multiple myeloma, lymphoid malignancies, Hodgkin lymphoma, non-Hodgkin lymphoma, and acquired immunodeficiency syndrome/human immunodeficiency virus-related lymphoma (Bae et al., 2011). CD138+ T cells have also been observed in systemic lupus erythematosus models (Liu et al., 2020). CD138 overexpression has been observed in many tumors, including breast, urinary bladder, gallbladder, pancreatic, ovarian, endometrial, and prostate cancers, making it an interesting therapeutic strategy for mAbs (Kind et al., 2019). In this study, we report the production and characterization of mQXL138A1 and study its in vivo efficacy and mechanism of action in various syngeneic mouse models.
Materials and Methods
Cell lines
All cell lines (except EMT-6) used in the in vivo tumor models were grown and maintained by Crown Bioscience Laboratories (Beijing, China). Neuro-2A and KNL-205 cells were cultured in Minimum Essential Media (MEM) medium supplemented with 0.01 nonessential amino acids and 10% fetal bovine serum (FBS). MC38, 4T1, Renca, Hepa 1–6, B16F10, and LLC1 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) medium supplemented with 10% FBS. MPC-11 cells were cultured in DMEM medium supplemented with 10% horse serum (HS). Clone M-3 was cultured in F-12K medium supplemented with 2.5% FBS and 15% HS. CT26, H22, B16BL6, Pan02, RM-1, and MBT2 cells were cultured in Roswell Park Memorial Institute (RPMI)1640 medium supplemented with 10% FBS. EMT-6 cells were purchased from American Type Culture Collection (Manassas, VA, USA) and cultured in Weymouth medium supplemented with 10% FBS. The B16-Blue cell line was purchased from InvivoGen (San Diego, CA, US) and cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 100 μg/mL normocin, and 2 mM
Vectors
The light and heavy chain variable regions, which together bind to murine CD138, were polymerase chain reaction (PCR)-amplified from a rat hybridoma-producing mAb 281-2 (Jalkanen et al., 1985). Total RNA was extracted using an RNeasy kit (Qiagen, Valencia, CA, USA), followed by cDNA synthesis with random hexamers using the Superscript III cDNA kit (Invitrogen, Carlsbad, CA, USA). PCR amplification of variable regions was performed, as described by Coloma MJ et al. (Coloma et al., 1992). The amplified variable regions were then cloned into mouse light chain and heavy chain expression vectors, as described by Trinh et al. (Trinh et al., 2013). For the heavy chain vector, murine IFN-α-1 was PCR-amplified from genomic DNA extracted from mouse spleen and fused to the C-terminus of murine IgG2a using an SGGGGS linker. A control heavy chain expression vector (mQXL138) that did not contain the murine IFN-α-1 gene was also constructed.
Mouse QXL138A1 fusion protein production and purification
The light and heavy chain expression vectors for anti-mCD138 mIgG2a fused to murine IFN-α (mQXL138A1) or anti-mCD138 mIgG2a (mQXL138) were transfected into the Chinese Hamster Ovary (CHO) cell line Pro5 using Lipofectamine LTX (Invitrogen) and plated into 96-well plates. After 12 days of selective pressure with 0.5 mM histidinol (Sigma, St. Louis MO, USA), cell culture supernatants from surviving transfectants were assayed for antibody expression by enzyme-linked immunosorbent assay (ELISA) (Trinh et al., 2013). Briefly, ELISA plates were coated with Fc-specific goat anti-mIgG (Sigma) antibody to capture secreted mIgG. After washing off any unbound protein, goat anti-mKappa light chain antibody-conjugated alkaline phosphatase (Southern Biotech, Birmingham, AL, USA) was used to detect bound mIgG. Clones were selected based on their absorbance intensities. Clones exhibiting the highest absorbance intensities were subcloned by limiting dilution and screened using ELISA.
Upon identification of an appropriate subcloned transfectant, cells were grown in Iscove’s modified Dulbecco’s medium (Gibco, Waltham, MA) supplemented with ultra-low bovine immunoglobulin G (IgG) FBS (Gibco) and expanded into roller bottles (Corning, Corning, NY, USA). Secreted murine IgG fusion protein was purified by affinity chromatography from harvested culture supernatants using protein A agarose (EMD Millipore, St. Louis, MO, USA). The purified protein was dialyzed against phosphate-buffered saline (PBS) supplemented with 2.5 mM ethylenediaminetetraacetic acid.
Fusion protein binding assay
B16-Blue (InvivoGen) cells were preincubated with Mouse Fc Block (Becton Dickinson, Franklin Lakes, NJ, USA) before incubation with 10 ug/mL of mQXL138 or mQXL138A1 for 1 h at 4°C in PBS supplemented with 2% bovine serum albumin (Sigma). Antibody binding was detected using PE-conjugated anti-mouse kappa light chain. Data were acquired using a Becton Dickinson FACSVerse flow cytometer (Becton Dickinson) and analyzed using FlowJo software.
B16-Blue IFN-α reporter assay
The B16-Blue IFN-α reporter cell assay was performed according to the manufacturer’s instructions (InvivoGen). On assay day 1, B16-Blue cells were harvested, rinsed, and resuspended in growth medium. In total, 180 μL of the cell suspension (∼75,000 cells) was added to each well of a 96-well plate. The cells were treated with titrated amounts of mouse IFN-α-1 (PBL Assay Science, Piscataway, NJ, USA) or mQXL138A1. To isolate the interaction between the IFN-α portion of mQXL138A1 and IFN-α receptor on the reporter cell line, CD138 antigen was blocked by preincubating the cells with 17.3 uM nonfusion mQXL138 for 5 h before adding mouse IFN-α-1 or mQXL138A1. The plates were then incubated at 37°C in a CO2 incubator for 24 h. The next day, the level of secreted embryonic alkaline phosphatase (SEAP) was measured using the QUANTI-Blue™ Solution (InvivoGen), and the optical density was measured using a plate reader (BioTek, Santa Clara, CA, USA) at 620 nM.
In vivo antitumor activity and toxicity in 15 syngeneic models
All animal care procedures were reviewed and approved by the Institutional Animal Care and Use Committee of CrownBio before execution and in accordance with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care. Tumor cells were inoculated subcutaneously into the flank region of 6- to 8-week-old female mice. Fifteen different tumor models were used in this study, and different mouse strains were used for different tumor models. For the Neuro-2A model, 1 × 106 cells were injected into A/J mice. For the MPC-11 model, 2 × 105 cells were injected into BALB/c mice. For the H22 model, 2 × 106 cells were injected into BALB/c mice. For the CT26, Renca, H22, and 4T1 models, 1 × 106 cells were injected into BALB/c mice. For the MBT-2 model, 4 × 106 cells were injected into the C3G/He mice. For the Hepa 1-6 model, 5 × 106 cells were injected into C57BL/6 mice. For the B16BL6 and B16F10 models, 2 × 105 cells were injected into C57BL/6 mice. For the Pan.02 model, 3 × 106 cells were injected into the C57BL/6 mice. For the LLC1 model, 3 × 105 cells were injected into C57BL/6 mice. For the KLN205 8 × 105 model, cells were injected into DBA/2 mice. For the Clon M-3 2 × 105 model, cells were injected into DBA/2 mice. Tumor volume (TV) was monitored until the group average was 80–100 mm3, and then, the mice were randomized into the vehicle or mQXL138A1 groups (n = 5). The day of randomization was defined as day 0. The vehicle group received PBS, and the treatment group received mQXL138A1 at 5 mg/kg at 10 μL/g through intravenous (i.v.) injections. Dosings were scheduled on days 1,3,7,10, 14, 17, 21, and 24 of the study. Different models received 4–8 doses based on the duration of the study. During the study, body weight (BW) and TV were measured 3 times per week. TVs were measured in 2 dimensions using calipers. TV was expressed in cubic millimeter using the formula V = (L × W × W)/2, where V is TV, L is tumor length (the longest tumor dimension), and W is tumor width (the longest tumor dimension perpendicular to L). During routine monitoring, the animals were checked for any effects of tumor growth and treatment on behavior, such as mobility, food and water consumption, and BW gain or loss. The percentage of BW change was calculated using the following formula: % Group Mean Change = mean (T-T0)/T0 × 100, where T is the current BW and T0 is the initial BW. The percentage of tumor growth inhibition (TGI) was calculated using the following formula: % ΔT/C = [mean(T) − mean(T0)]/[meanI − mean(C0)] × 100%, where T is the current TV, T0 is the current group initial TV, C is the control group TV, and C0 is the initial TV of the control group. The animals remained in the study until the individual TV exceeded 3000 mm3 or the mean TV in a group exceeded 2000 mm3. The study for each model was terminated on the day that the last 5 animals in the vehicle group were alive.
Flow cytometry detection of CD138 surface expression
Tumor cells 1 × 106 were harvested and resuspended in 100 μL staining buffer (PBS + 2% FBS) with 1 μg/mL Fc-Block (BD, CAT# 553141). The cells were incubated at 4°C in the dark for 10 min. T cells were then stained with PE-conjugated mouse anti-CD138 antibody (BioLegend, CAT# 142503) or rat IgG2a kappa (BioLegend, CAT#142503) isotype control for 30 min at 4°C in the dark. The cells were washed and stained with PerCP-conjugated 7-ADD dye (BioLegend, CAT# 420403). The samples were analyzed using a flow cytometer (Beckman Coulter, Brea, US). Data were analyzed using Kaluza software (Beckman Coulter, Brea, CA, US). The Mean fluorescence intensity (MFI) of CD138+ viable cells was calculated. The fold changes in MFI compared with their isotype control were categorized as follows: (+ 1–10-fold, ++ 10–100-fold, +++ 100–1000-fold, and ++++ >1000-fold).
In vivo efficacy and flow cytometry detection of tumor infiltration lymphocytes in EMT-6 model
For the efficacy study, 5 × 105 EMT-6 cells were injected into the BALB/c mice (n = 8). The vehicle group received PBS, and the treatment group received mQXL138A1 at 5 mg/kg through i.v. injections on days 8, 10, 13, and 17. Bidirectional tumor growth was measured throughout the experiment, as described earlier, and the mice were sacrificed when the tumors reached 1.4 cm.
For the assessment of tumor infiltration lymphocytes (TILs), 5 × 105 EMT-6 cells were inoculated subcutaneously into the rear flanks of BALB/c mice. Animals were randomized into the vehicle and mQXL138A1 groups (n = 5). The vehicle group received PBS, and the treatment group received mQXL138A1 at 5 mg/kg at 10 μL/g through i.v. injections, which were administered i.v. on days 18, 19, and 20 post-tumor implantation, and the tumors were harvested on day 22. The tumors were cut into small pieces, transferred to the tubes containing the digestion buffer and incubated at 37°C for 25 min while shaking. The tumors were passed through a 70 μm strainer and washed twice with RPMI medium. Red blood cells (RBCs) were lysed using an RBC lysis buffer, and the samples were washed with cold PBS. Cells were counted and resuspended in flow cytometry staining buffer (eBioscience, CAT# 004–4222-26). Before staining, samples were treated with anti-mouse CD16/32 (eBioscience CAT# 14016185) for Fc blocking. Cells were stained with fixable viability dye at 4°C for 30 min in the dark. The cells were washed, stained with antibodies, and incubated at 4°C for 30 min in the dark. The samples were washed with flow cytometry buffer and analyzed using Attune NxT (ThermoFisher Scientific, Waltham, MA, USA). The list of antibodies and gating strategies is summarized in Supplementary Table S2.
Statistical analysis
GraphPad software was used to statistically analyze differences between groups. All tests were 2-tailed and conducted at 95% confidence interval. To compare TV of different groups on a specified day, Bartlett’s test was used to check the assumption of homogeneity of variance across all groups. If the P value of Bartlett’s test was ≥0.05, then one-way analysis of variance (ANOVA) was used to test overall equality of means across all groups. If the P value of the one-way ANOVA <0.05, further post hoc testing was performed by Tukey’s honest significant difference tests for all pairwise comparisons or Dunnett’s tests for comparing each treatment group with the vehicle group. If the P value of Bartlett’s test was <0.05, the Kruskal–Wallis test was used to test the overall equality of medians among all groups. If the P value of the Kruskal–Wallis test was <0.05, then post hoc tests were performed by Conover’s nonparametric test for all pairwise comparisons or for comparing each treatment group with the vehicle group; both results were under P value adjustment.
Results
Production and characterization of mQXL138A1
mQXL138A1 consists of a murine anti-mouse CD138 IgG2a genetically fused to mIFN-α-1 at the C-terminus of each heavy chain (Fig. 1A). Cotransfection of the light and heavy chain vectors for mQXL138 or mQXL138A1 into CHO cells, followed by limiting dilution subcloning, generated cells secreting proteins that were purified using protein A agarose affinity chromatography. Nonreduced sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) revealed an apparent size difference between intact mQXL138 and mQXL138A1 resulting from IFN fusion. Reduced SDS-PAGE confirmed the expected increase in mass associated with mQXL138A1 (Fig. 1B). The binding of mQXL138 and mQXL138A1 to mouse CD138 was assessed by flow cytometry using CD138-positive B16-Blue melanoma cells. The parental antibody and mQXL138A1 both bound to B16-Blue cells with mQXL138A1, providing a slightly higher MFI, potentially demonstrating additional binding owing to IFN-α receptor interaction (Fig. 1C).
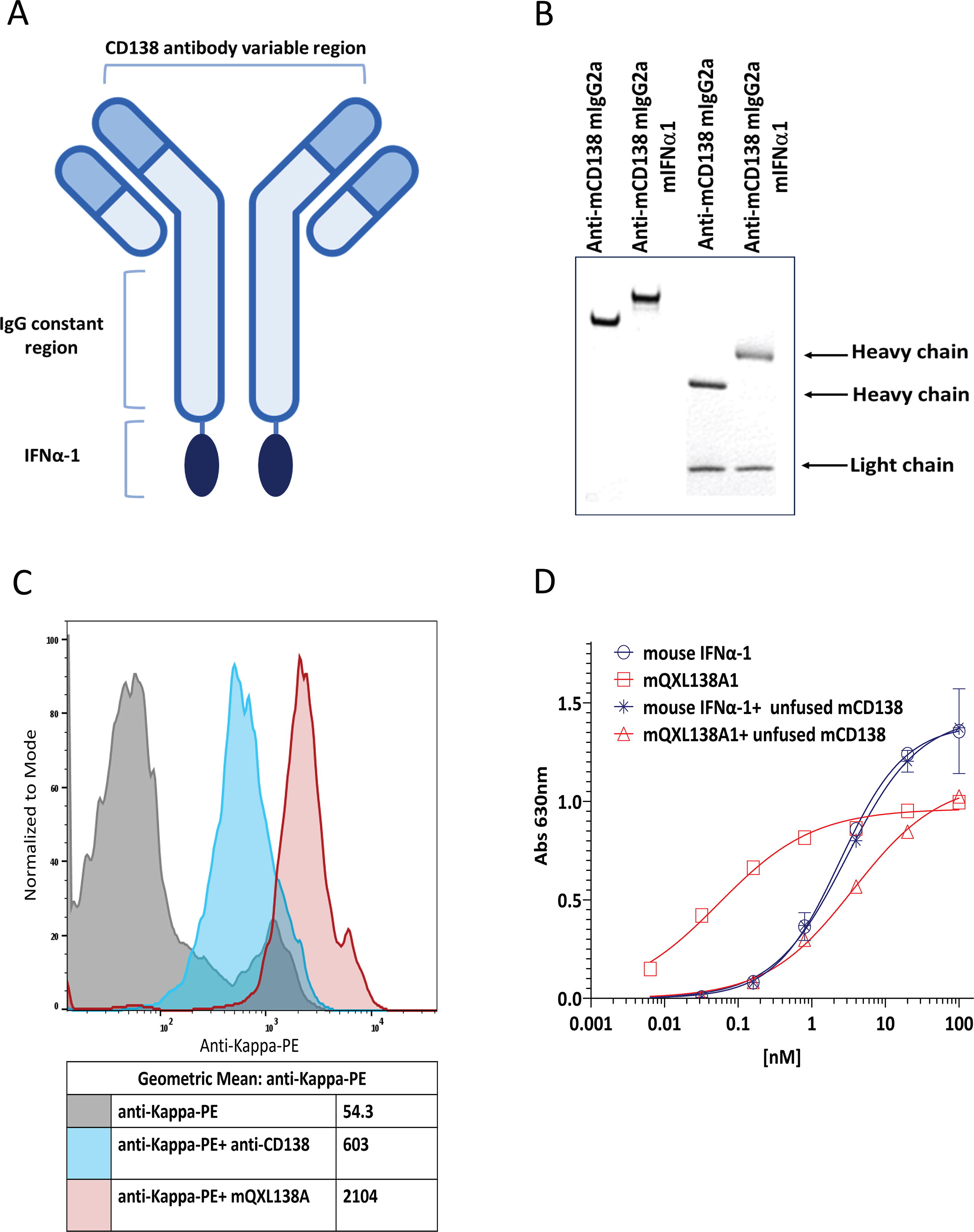
mQXL138A1 demonstrates IFN-α-dependent activity in vitro
B16-Blue IFN-α/β reporter cells (InvivoGen) were used to evaluate the IFN-α receptor activation. They were derived from the murine B16 melanoma cell line of C57BL/6 origin after stable transfection with a SEAP reporter gene under the control of the IFN-α/β-inducible ISG54 promoter, which was enhanced by a multimeric Interferon-sensitive response element (ISRE). B16-Blue IFN-α/β cells allow for the detection of bioactive murine type I IFNs by monitoring the activation of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) and IFN-stimulated gene factor 3 (ISGF3) pathway. IFN-β and all the IFN-α subtypes bind to a heterodimeric transmembrane receptor of the cell line composed of the subunits IFNAR1 and IFNAR2, which are associated with the tyrosine kinases Tyk2 and Jak1 (Janus kinase 1), respectively. These kinases phosphorylate STAT1 and STAT2, which then dimerize and interact with IFN regulatory factor 9 (IRF9), leading to the formation of the ISGF3 complex and production of SEAP, which is then quantitated using the detection medium QUANTI-Blue Solution (InvivoGen) (Supplementary Fig. S1) (InvivoGen, 2024). B16-Blue IFN-α/β reporter cells were treated with either wild-type mouse IFN-α-1 or mQXL138A1 in the presence or absence of nonfusion mQXL138. IFN-α signaling was measured after overnight incubation. The results showed IFN-α activity in mQXL138A1 with EC50 = 0.1 nM and showed lower EC50 than wild-type mouse IFN-α-1 (EC50 = 2.4 nM). In addition, blocking the CD138 antigen (with mQXL138) reduced the potency of mQXL138A1 by 30-fold (EC50 = 3 nM) (Fig. 1D). Together, these results demonstrate that targeting IFN-α to the cell surface enhances its potency by 24- to 30-fold.
Treatment with mQXL138A1 induces TGI in different cancer models independent of the CD138 surface expression level
The in vivo antitumor efficacy of mQXL138A1 was tested in 15 subcutaneous syngeneic mouse tumor models. The animals were inoculated with the tumor cells, as described in the Materials and Methods section, and randomized into the treatment and vehicle groups when the TV reached 80–100 mm3 counted as day 0. Dosing started on day 1, and animals received 4–8 doses on days 1,3,7,10, 14, 17, 21, and 24 based on the tumor growth rate and length of the study. Details regarding dosing numbers and frequencies can be found in Supplementary Table S1. The mean TGI was calculated in comparison with that of the vehicle group on the last day that all animals in the vehicle group remained on the study. The surface expression of CD138 was measured using flow cytometry. Treatment with mQXL138A1 inhibited tumor growth in all models. The percentage of tumor growth inhibition was different in each model and was independent of CD138 surface expression and tumor indication. Significant tumor growth inhibition (P < 0.05) was observed in all models except KLN-205, CT26, Hepa 1-6, and Pan02 (Table 1). The median %TGI was 62%, and the highest %TGI was observed in MC38 colon and H22 liver models. When comparing the level of CD138 surface expression with TGI, no direct correlation was found, as there were models with higher levels of CD138 expression but lower TGI, such as LLC1, and there were models with lower levels of CD138 expression but higher TGI, such as MC38 (Table 1, Fig. 2).

Antitumor efficacy of mQXL138A1 was independent of CD138 expression: The study was conducted, as described in Table 1. The graphs show the tumor volumes in different models with different levels of CD138 expression (+++ H22, ++ LLC1, + MC38, and +++ MPC11), showing the tumor growth inhibition was independent of CD138 expression. The P values are indicated by * as *** is P < 0.01 and ** is P < 0.05.
Antitumor Efficacy of mQXL138A1 in Various Subcutaneous Tumor Models
Tumor cells were injected subcutaneously in the flank of the mice. Animals were randomized into the vehicle and QXL138A1 groups (n = 5) when tumors reached 80–100 mm3 (day 0) and were dosed via i.v. injections. Tumors were measured 3 times a week. TGIs and P values in different models were calculated in comparison with the vehicle groups on the day all animals were alive in the vehicle group. The + were calculated by fold changes of MFI compared with their isotype control (+: 1–10-fold, ++: 10–100-fold, +++: 100–1000-fold, and ++++: >1000-fold). The treatment-related rate of mortality shows in the death column the number of animals found dead during the study.
TGI, tumor growth inhibition.
Treatment with mQXL138A1 induces low level of toxicity in mouse
The tolerability of mQXL138A1 was also assessed during the efficacy studies in the 15 syngeneic tumor models. The BW of the animals were measured 3 times per week, and they were observed for any sign of clinical toxicity, such as weight loss, behavioral changes, mobility, fur conditions, and food and water consumption. The percentage of BW change was calculated, as described in the Materials and Methods section. Treatment with mQXL138A1 resulted in weight loss and mortality in some models. No mortality or minor BW loss (<5%) was observed in 8 of 15 models (MPC-11, LLC1, B16BL6, B16F10, Pan02, RM-1, KLN205, and Clone M-3). Treatment-related BW loss was observed in the following models: minor BW loss was observed in CT26 and Hepa1-6. Moderate BW loss (5%−10%) was observed in the Neuro-2A, Renca, and H22 models. Severe BW loss (19%) was observed only in the MBT-2 model (Supplementary Table S1). Treatment-related deaths were observed in CT26, Renca, H22, Hepa1-6, and MBT-2 models (Table 1). Of the 75 animals treated with either vehicle or mQXL138A1, 14 animals treated with mQXL138A1 and 4 animals treated with the vehicle died during the dosing period (Supplementary Table S1).
mQXL138A1 induces TGI in the EMT-6 breast cancer model by enhancing cytotoxic T cell infiltration and inducing immune cell activation in the tumor microenvironment
To further understand the mechanisms by which mQXL138A1 induces antitumor effects, the antitumor efficacy and effect of mQXL138A1 on the population of TILs were evaluated in EMT-6-bearing mice. To evaluate antitumor efficacy, BALB/c mice were inoculated with EMT-6 breast cancer cells and treated with the vehicle control or mQXL138A1 at 5 mg/kg on days 8, 10, 13, and 17 after tumor injection. Compared with the vehicle group, treatment with mQXL138A1 inhibited tumor growth in the EMT-6 model (Fig. 3A).
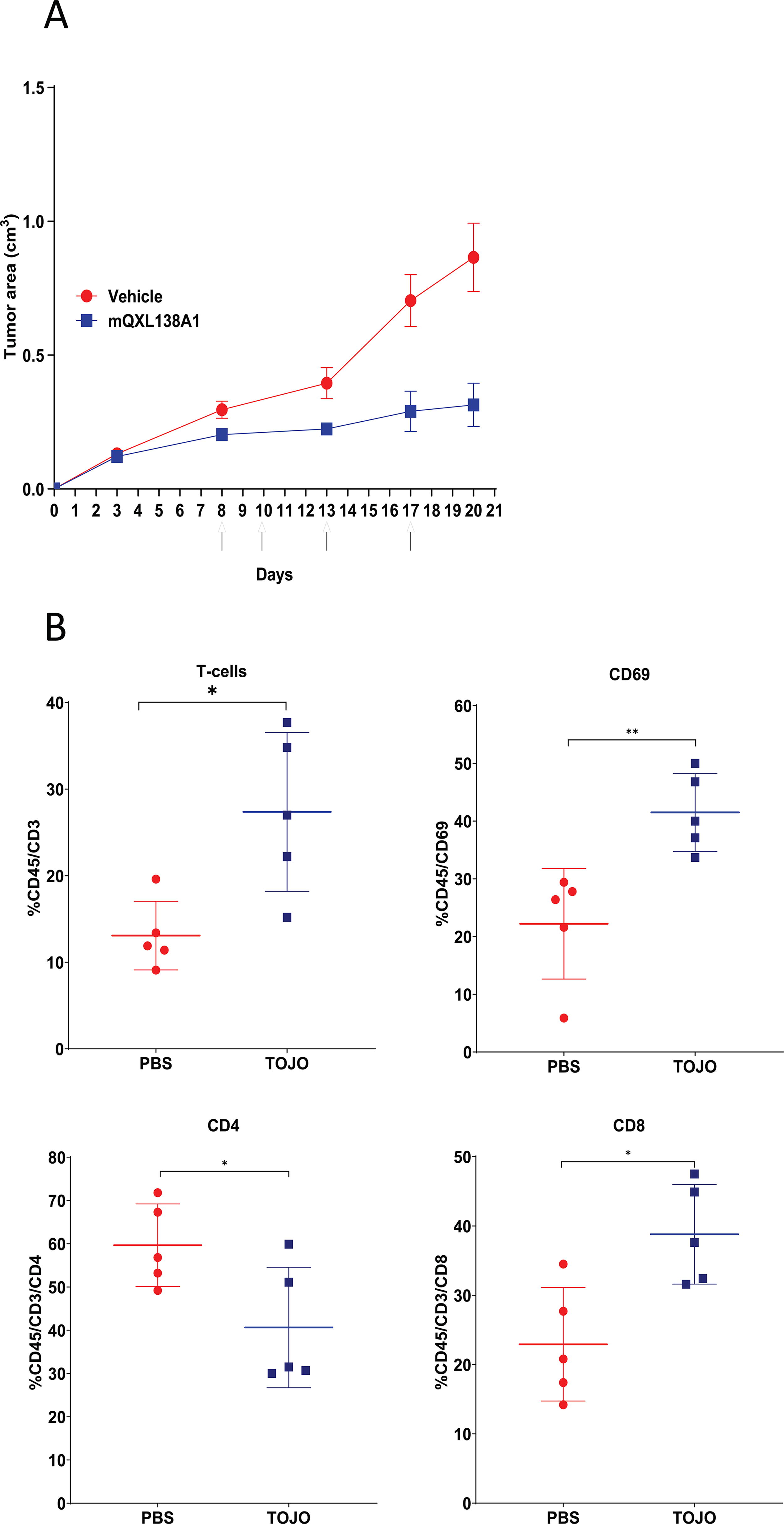
To analyze the TIL population, EMT-6 tumor-bearing mice were i.v. injected with 5 mg/kg mQXL138A1 on days 18, 19, and 20 post-tumor implantation, and the tumors were harvested on day 22. The tumors were harvested and dissociated into single cells, and the lymphocyte population was analyzed by staining the cells and assessing them using a flow cytometer. The results showed an increase in the percentage of CD69+ cells among the viable CD45+ cells compared with that in the PBS-treated group, which is an indication of lymphocyte activation. When compared with the PBS group, the tumors from the mQXL138A1-treated mice had a higher population of cytotoxic T cells (CD45+/CD3+/CD8+), which can induce an antitumor immune response in the tumor microenvironment. A lower population of T helper cells (CD45+/CD3+/CD4+) was also observed in the mQXL138A1-treated group, which may be due to the immunostimulatory properties of mQXL138A1, resulting in a decrease in the population of regulatory CD4+ cells (Fig. 3B).
Discussion
Cytokines play a crucial role in the regulation of immune responses and are potential therapeutic agents for various cancers. Among cytokines, IFN-α has been used widely for the treatment of different hematological malignancies and solid tumors, such as melanoma, renal carcinoma, and Kaposi’s sarcoma (Belardelli et al., 2002; Kirkwood, 1998), and its translation into clinical practice faces challenges, including significant toxicities. These issues can arise from the pleiotropic nature of cytokines and off-target cell activation (Bonati and Tang, 2021; Jia et al., 2016; Kirkwood, 2002). Previous efforts to improve the therapeutic window have focused on the treatment of hematological malignancies [13] (Trinh et al., 2013; Xuan et al., 2010). This study demonstrates, for the first time, that tumor-targeted IFN-α can have broad activity in multiple solid tumor indications.
The results showed that mQXL138A1 maintained its ability to bind to CD138 and IFN-α receptor-expressing cell lines, allowing targeted delivery of the cytokine to CD138-expressing tumor cells. mQXL13A1 showed in vitro activity in a mouse IFN-α reporter cell line and exhibited significant antitumor efficacy in various syngeneic mouse models. Inhibition of tumor growth by mQXL138A1 has been observed in myeloma, neuroblastoma, colon, renal, liver, lung, melanoma, prostate, and bladder cancer models. The extent of tumor growth inhibition by mQXL138A1 was independent of the CD138 surface expression level of the tumor cells used to develop the tumor models.
The efficacy of mQXL138A1 in mouse tumor models may be driven by altering the immune environment within the tumor. The analysis of tumor-infiltrating lymphocytes in this study showed that mQXL138A1-induced lymphocyte activation increased the frequency of CD8+ T cells and decreased the population of CD4+ T helper cells. CD8-expressing cytotoxic T cells are known to be among the most powerful effector cells in anticancer immune responses, as they exhibit cytotoxicity against tumor cells (Raskov et al., 2021). Exhaustion and dysfunction of cytotoxic T cells have been observed in cancer owing to the complex immunosuppressive features of the tumor microenvironment. The presence of regulatory T cells is an immunological barrier that affects CD8+ T cell-mediated antitumor immune responses and induces exhaustion. Treatments that can activate and expand cytotoxic T cells in the tumor environment and eliminate immunosuppressive immune cell populations may support a durable and effective immune response (Hope et al., 2020; Penaloza-MacMaster, 2017; Zhang et al., 2020). This study encounters a limitation in data availability on the subpopulation of CD4+ T cells to identify the specific phenotype that decreases in the tumor microenvironment. To address this gap, further analyses are required to investigate the phenotypes of CD4+ TILs in detail. Further analysis should be performed to determine whether treatment with mQXL138A1 can enhance functional markers of CD8+ T cells, such as IFN-γ secretion or granzyme expression.
Overall, systemic toxicity was occasionally observed when mice were treated with mQXL138A1. Less than 10% BW loss was observed in some syngeneic models, and major BW loss was observed only in the MBT-2 model at the end of the study. Approximately 18.7% of animals treated with mQXL138A1, and 5.3% of animals treated with the vehicle died before being removed from the study owing to tumor growth, suggesting treatment-related toxicity. To further decrease the systemic toxicity observed in some of the models, our team has developed a new masking technology, whereby a peptide mask is attached to the C-terminus of the cytokine via a tumor-selective protease-cleavable linker (data not published). This structure allows conditional cytokine activation, where the masked cytokine is preferentially activated (“demasked”) by proteases in the tumor microenvironment. The use of mQXL1381 as a murine surrogate made it possible to study the molecule in an extensive number of mouse tumor models, confirming its efficacy and functionality and supporting additional clinical and more selective studies in human models and clinical studies.
Conclusion
Overall, this study demonstrated that the systemic administration of mQXL138A1 has efficacy in solid tumor indications and multiple myeloma syngeneic mouse tumor models. Although IFN-α has shown promise in treating various malignancies, its widespread clinical use has been hindered by significant toxicities attributed to its pleiotropic nature. This research presents an approach using tumor-targeted IFN-α-1 (mQXL138A1), which demonstrated potent antitumor activity in syngeneic mouse models. The efficacy of mQXL138A1 was associated with alterations in the tumor immune microenvironment, characterized by enhanced activation of cytotoxic CD8+ T cells and suppression of regulatory T cells. Despite occasional systemic toxicity observed in preclinical models, ongoing efforts to mitigate these effects through innovative masking technologies hold promise for enhancing the safety profile of mQXL138A1. This study provides compelling evidence to support further preclinical investigation of mQXL138A1 in additional cancer models, with the potential to advance the development of more selective and efficacious immunotherapeutic anticancer strategies.
Authors’ Contributions
T.S.: Conceptualization, methodology, validation, formal analysis, resources, data curation, writing—original draft, visualization, and project administration. K.R.T.: Conceptualization, methodology, validation, formal analysis, investigation, resources, data curation, writing—original draft, writing—reviewing and editing, and visualization. A.V.: Conceptualization, methodology, validation, investigation, resources, data curation, writing—reviewing and editing, and visualization. S.L.M.: Conceptualization, methodology, supervision, writing—reviewing and editing, project administration, and funding acquisition. D.R.S.: Conceptualization, methodology, supervision, writing—reviewing and editing, project administration, and funding acquisition.
Footnotes
Authors Disclosure Statement
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in, or financial conflict with, the subject matter or materials discussed in the article, apart from those disclosed.
Funding Information
This study was supported and funded by Nammi Therapeutics.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2