
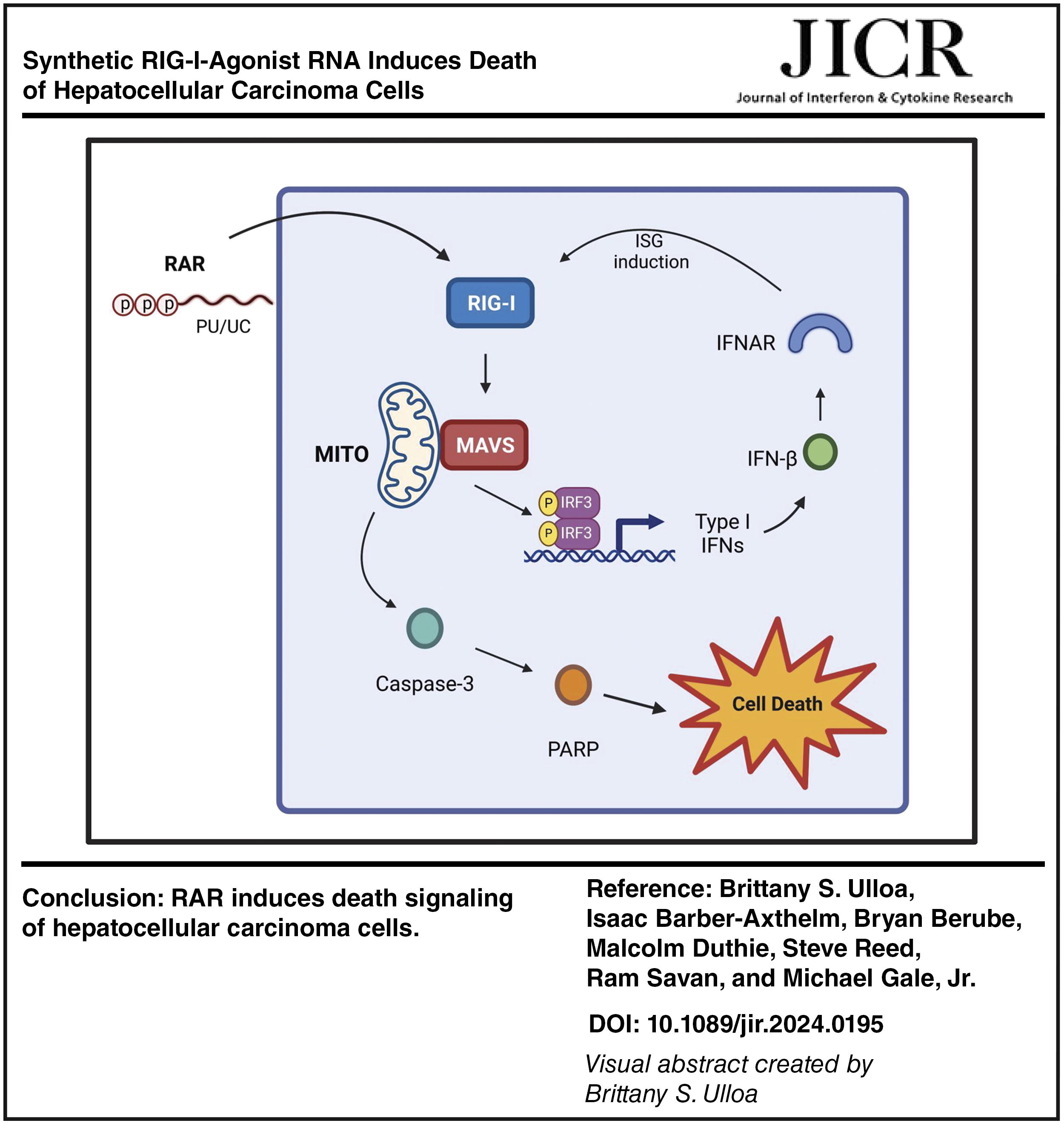
Abstract
Retinoic acid-inducible gene I (RIG-I) is a critical sensor of viral RNA and is activated in response to binding to RNA containing exposed 5’-triphosphate (5’ppp) and poly-uridine to trigger innate immune activation and response including induction of type I and III interferons (IFNs). RIG-I signaling plays a key role in not only restricting RNA virus infection but also suppressing tumor progression via oncolytic signaling. We evaluated the actions of a specific RIG-I agonist RNA (RAR) as a potential therapeutic against model tumor cell lines representing hepatocellular carcinoma (HCC). RAR constitutes a synthetic-modified RNA motif derived from the hepatitis C virus genome that is specifically recognized by RIG-I and induces innate immune activation when delivered to cells. We found that RAR directs RIG-I-dependent signaling to drive HCC cell death. Analysis of knockout cell lines lacking RIG-I, mitochondrial activator of virus signaling, or IRF3 confirmed that RAR-induced cell death signaling propagates through the RIG-I-like receptor (RLR) pathway to mediate caspase activation and HCC cell death. RAR-induced cell death is potentiated by type I IFN. Thus, RAR actions trigger HCC cell death through RIG-I linkage of RLR, caspase, and IFN signaling programs. RAR offers a potent application in antitumor therapeutic strategies leveraging innate immunity against liver cancer.
Introduction
In vertebrates, the intracellular innate immune response to virus infection is triggered by pathogen recognition receptors (PRRs). Retinoic acid inducible gene I (RIG-I) is the charter member of the RIG-I-like receptor (RLR) family of cytosolic PRRs that recognize RNA virus infection to trigger the innate immune response (Kell et al., 2015; Kell and Gale, 2015; Pattabhi et al., 2016; Saito et al., 2008; Yoneyama et al., 2004). RLRs signal through the mitochondrial activator of virus signaling (MAVS) adaptor protein to direct downstream activation of interferon regulator factor 3 (IRF3), nuclear factor-kappa B (NF-KB), and other transcription factors and their target genes, including types I and III interferons (IFNs), to induce an antiviral state that can limit virus replication and spread through direct antiviral actions and cell death signaling (Iurescia et al., 2020; Pattabhi et al., 2016; Ren et al., 2020; Saito et al., 2008). IFN and other cytokine products of RLR signaling also serve to program innate immune cells for activating and polarizing the adaptive immune response (Kell et al., 2015; Kell and Gale, 2015; Pattabhi et al., 2016; Saito et al., 2008). RIG-I is expressed in most cell types, including cancer cells, at a low level and is induced to higher levels in response to IFN (Elion and Cook, 2018; Kell and Gale, 2015; Saito et al., 2008). While RIG-I plays an essential role to program immunity against most RNA viruses (Kell et al., 2015; Kell and Gale, 2015; Pattabhi et al., 2016; Saito et al., 2008), an increasing number of studies show that it can also direct antitumor actions triggered through RLR signaling to suppress different cancer types, including melanoma, acute myeloid leukemia, and cervical cancer (Castiello et al., 2019; Cattaneo et al., 2008; Cui, 2017; Das et al., 2019; Duewell et al., 2014; Elion et al., 2018; Elion and Cook, 2018; Grützner et al., 2024; Helms et al., 2019; Iurescia et al., 2020; Javanmard et al., 2020; Kaneda, 2013; Li et al., 2021). In cancer cells, the two major outcomes of RIG-I signaling are type I IFN production and programmed cell death (Cui, 2017; Elion and Cook, 2018; Kaneda, 2013). Problematically, tumor cells impose various strategies to evade immunosurveillance (Bonaventura et al., 2019) and establish an immunosuppressive tumor microenvironment (TME) that fosters cancer progression (Bonaventura et al., 2019; Grützner et al., 2024; Li et al., 2021; Piya et al., 2011; Ren et al., 2020). Type I IFN can contribute to spontaneous activation of antitumor T-cell responses, highlighting the innate immune actions as a strategy for controlling cancer progression (Elion and Cook, 2018; Piya et al., 2011). Thus, engaging innate immunity by targeting RIG-I and the RLR pathway to mediate innate immune activation offers a promising approach in immunotherapeutic strategies against cancer that can overcome the immunosuppressive TME.
Hepatocellular carcinoma (HCC) is among the most common malignancies and causes of cancer-related deaths (Brown et al., 2023). Increasingly, the incidence of HCC is linked with obesity and related pathology including fatty liver disease and liver cirrhosis (Forner et al., 2018). Moreover, persistent infection by hepatitis C virus (HCV) or hepatitis B virus continues to be a major etiology of HCC worldwide (Dash et al., 2005; Levrero and Zucman-Rossi, 2016). Treatments for HCC historically have focused on systemic therapies that slow cancer progression wherein just a fraction of patients obtain long-term benefit. Targeted therapeutics against HCC show promise for clinical benefit (Yang et al., 2023). Targeting HCC solid tumors and tumor cells for suppression and cell death presents an attractive strategy to limit tumor progression and enhance antitumor immunity (Yang et al., 2023). For example, RIG-I expression level (Zhou et al., 2020) and activity (Zao et al., 2021) have been linked with death of HCC cells and M1 immune polarization, implying that strategies to target and activate RIG-I in the HCC tumor cell can offer an immunotherapeutic approach, alone or in combination, to treat HCC.
Several RIG-I or RLR-agonists have been shown to selectively induce apoptosis of human cancer cell lines and model tumors without triggering cell death in immortalized or primary cells (Brägelmann et al., 2021; Glas et al., 2013; Grützner et al., 2024; Kurooka and Kaneda, 2007; Li et al., 2017; Sugii et al., 2021; Zheng et al., 2021). Fragments of genomic viral RNA can selectively promote apoptosis in cancer cells via the induction of proapoptotic genes including tumor necrosis factor-related apoptosis-inducing ligand and NOXA, downstream of the RIG-I/MAVS pathway (Castiello et al., 2019; Helms et al., 2019; Iurescia et al., 2020; Kaneda, 2013). Mechanisms by which RIG-I triggers programmed cell death include activation of the intrinsic apoptotic pathway, the extrinsic apoptotic pathway, and pyroptosis (Castiello et al., 2019; Duewell et al., 2014; Duprez et al., 2009; Koren and Fuchs, 2021; Orning and Lien, 2021). Intrinsic apoptosis is regulated by the proapoptotic BCL2 family members (e.g., NOXA and PUMA) and results in permeabilization of the mitochondrial outer membrane (Kale et al., 2018; Piya et al., 2011; Ploner et al., 2008). Subsequent leakage of apoptosis-inducing proteins, such as cytochrome c, out of the mitochondria then activates the initiator caspase-9 resulting in a caspase cascade that concludes with cellular apoptosis (Chattopadhyay et al., 2010; Kaneda, 2013). Extrinsic apoptosis involves engagement of a death receptor by its cognate death ligand, resulting in the activation of initiator caspase-8 or -10 which then activates effector caspases-3, -6, and -7, effectively executing cellular apoptosis (Duprez et al., 2009; Koren and Fuchs, 2021; Orning and Lien, 2021). RIG-I signaling can also induce pyroptosis, where RIG-I engagement activates the inflammasome by forming a protein complex containing apoptosis-associated speck-like proteins and caspase-1 (Bergsbaken et al., 2009; Fink and Cookson, 2005; Wu et al., 2019). Activated caspase-1 promotes gasdermin D translocation to the plasma membrane to form membrane pores, causing cellular swelling and lysis. In addition to apoptosis and pyroptosis, an alternate form of caspase-independent programmed cell death is necroptosis (Gong et al., 2019; Grootjans et al., 2017; Kist and Vucic, 2021; Liu and Jiao, 2019; Schock et al., 2017). Necroptosis can be initiated by multiple stimuli including but not limited to TNF or by TLR3/TLR4 ligands, DNA damaging agents, and T-cell receptor ligation (Fink and Cookson, 2005; Grootjans et al., 2017; Liu and Jiao, 2019). Signaling in cells deficient of the death receptor adapter protein Fas-associated death domain (FADD) or caspase-8 leads to deubiquitination of the serine/threonine kinase, RIP1, and its recruitment to family member, RIP3, to form a “necrosome” complex (Grootjans et al., 2017; Yoon et al., 2017). Cell death programs can be blocked by treatment of cells with the pan-caspase inhibitor zVAD-FMK (zVAD) (Li et al., 2019).
Here we show that synthetic RIG-I agonist RNA (RAR), representing a modified 100 nucleotides (nt) RIG-I pathogen associated molecular pattern (PAMP) motif of the HCV RNA genome (Saito et al., 2008; Schnell et al., 2012) containing an exposed triphosphate motif, promotes in vitro death of Huh7 cells and HepG2 HCC cells. Addition of the pan-caspase-inhibitor, zVAD, ablates cell death, revealing that RAR directs a form of caspase-mediated cell death in parallel with RIG-I mediated innate immune activation, with resulting IFN production and signaling potentiating RAR- mediated cell death. These findings inform specific applications for further consideration of RAR as a possible component of HCC tumor therapy.
Oligonucleotide Primer Sequences
Materials and Methods
Cells and culture methods
Huh7 and HepG2 cells were obtained from the American Type Culture Collection. All cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS), 2 mM
RAR and XRNA
RAR is in preclinical development for applications as a therapeutic agent. RAR and RNA motif form the hepatitis C virus genome RNA X region (XRNA) have been extensively described and the sequence of each is reported (Saito et al., 2008; Schnell et al., 2012). RAR and XRNA were generously supplied by HDT Bio Corp. Both were generated from template DNA using T7 polymerase. All RNA synthesis reagents were kept on ice but assembled at room temperature. DNA template was produced from oligonucleotides as described (Saito et al., 2008; Schnell et al., 2012). Oligonucleotides were annealed using a heat block incubation in normal saline starting at 95°C for 5 min, dropping 1°C every 30 s until reaching 4°C. RNA was purified by phenol-chloroform extraction, ethanol precipitation, and size-exclusion chromatography. RNA products were verified by gel electrophoresis and imaging.
RAR sequence: 5’ GGCCAUCCUGUUUUUUUCCCUUUUUUUUUUUCUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUCUCCUUUUUUUUUCCUCUUUUUUUCCUUUUCUUUCCUUUU 3’
XRNA sequence: 5’ UAAUACGACUCACUAUAGGUGGCUCCAUCUUAGCCCUAGUCACGGCUAGCUGUGAAAGGUCCGUGAGCCGCUUGACUGCAGAGAGUGCUGAUACUGGCCUCUCUGCAGAUCAAGU 3’
RAR transfection
RAR and XRNA (negative control) were transfected using the Mirus TransIT-mRNA Transfection Kit. TransIT-mRNA and mRNA-Boost reagents were warmed to room temperature and gently vortexed. Next, 100 μL of Opti-MEM Reduced-Serum Medium was placed in sterile tubes. We then added 1 μg (1μL of a 1μg/μL stock) RNA and gently pipetted to mix. We also added 1 μL mRNA Boost Reagent and 1 μL of TransIT-mRNA and pipetted gently to mix. Samples were gently flicked 15 times and subjected to a quick 2 s spin. Samples were then incubated at room temp for 2–5 min to allow for complexes to form. MOCK samples were prepared with TFXs only (no RAR or XRNA). The complexes were then distributed to the cells in complete growth medium. The vessel was rocked back and forth gently to evenly distribute the transfection complexes. Samples were incubated for 4–48 h depending on the nature of the goal of each experiment. Cells were harvested and assayed as required.
Staurosporine treatment
Staurosporine (Millipore) was used as a cell death control. Staurosporine was prepared at concentrations of 0.5, 1, 5, and 10 μM in supplemented DMEM. Cells were seeded the prior day, and staurosporine was administered by replacing the media with prepared staurosporine media. Cell death was then monitored via incucyte.
Immunoblot
Cells were lysed in RIPA buffer (Sigma) with freshly added protease inhibitor cocktail (Sigma), phosphatase inhibitor cocktail (Millipore), and Okadaic acid (Thermo). Lysates were separated on 8% Bis-Acrylamide sodium dodecyl sulfate (SDS) gels and transferred onto Polyvinylidene fluoride (PVDF) membranes (Millipore), then blocked/probed overnight in 4°C incubation with primary antibody diluted in 4% BSA blocking buffer. Membranes were stained overnight with the following primary antibodies diluted in blocking buffer: RIG-I (no. AG-20B-0009; 1:1000; AdipoGen), MAVS (A300-782A; 1:1000; Bethyl), IRF3 (no. 4302; 1:1000; Cell Signaling Technology), pIRF3 S386 (no. 37829; 1:1000; Cell Signaling Technology), IFIT1 (no. 971; 1:1000; in-house), Actin (no. mab1501; 1:1000; Millipore), PARP (no. 9542; 1:1000; Cell Signaling Technology), caspase-3 (no. 9668; 1:1000; Cell Signaling Technology), cleaved caspase-3 (no. 9664; 1:1000; Cell Signaling Technology). After washing, membranes were incubated for 1 h at room temperature with HRP-conjugated secondary antibodies diluted in TBS-tween. Membranes were incubated in ECL (Fisher Scientific) and detected using a ChemiDoc XRS+ System (Bio-Rad).
Cell death assays
HCC cells were seeded at a concentration of 1 × 105 cells per well in 24-well dishes (Corning). Cells were then transfected 16 h post seeding with MOCK, XRNA, or RAR 400 ng/mL. Cells were then stained with the cell permeable dye Sytox green 1:1000, 4 h post transfection. PAMP-induced cell death was assessed in real time via quantification of Sytox dye uptake through live cell imaging using an incucyte instrument over a 24- to 50-h period. Percent cell death was calculated as a ratio of dead cells (Sytox) over live cells (Syto) and monitored over time.
Cell titer glo viability assays
HCC cells were seeded at a concentration of 1.5 × 104 cells per well in 100 mL complete DMEM, in 96-well dishes (Corning). Cells were then transfected 16 h post-seeding with MOCK, XRNA (400 ng/mL), RAR (100, 200, or 400 ng/mL), or treated with Stuaurosporine (5 μM). Plates were incubated for 72 h. Cell Titer Glo 2.0 Reagent (100 μL, Promega) was then added to each well using a multichannel pipette. The contents were mixed in each well by pipetting gently up and down 4–5 times. Once all wells were mixed, the contents of each well was transferred to new white opaque 96-well plates (Thermo Fisher). Samples were allowed to incubate at room temperature for 10 min to stabilize the luminescence signal. Luminescence was read at an integration time of 0.1 s. Relative luminescence was calculated as a percent ratio according to the transfection (TFX) mock samples.
qRT-PCR
Total RNA was isolated from cell lysates using the RNeasy Kit (QIAGEN) and digested with DNase I (QIAGEN) on column. We isolated 200 ng total RNA, and this was subjected to cDNA synthesis using the iScript cDNA Synthesis Kit (BioRad). We then diluted cDNA1:4 in nuclease-free water, and qPCR was performed using SYBR Select Master Mix (Thermo) and gene specific primers on the ABI 7500 Real-Time PCR System.
Interferon treatment
Human IFN-β (Toray) was prepared in complete supplemented DMEM at concentrations of either 10, 100, or 1000 U per mL. This was then used to replace the media on Huh7 or HepG2 cells that had been seeded the day before (about 16 h before). Cells were stained with Syto and Sytox 4 h posttreatment, and cell death was monitored over time via incucyte. For interferon pretreatments, interferon was administered as stated above, followed by RAR transfection 24 h later.
zVAD-FMK treatment
To interrogate cell death signaling, zVAD-FMK (Sigma) was used as a pan-caspase inhibitor. zVAD-FMK was prepared at a concentration of 100 μM in supplemented DMEM. Cells that had been seeded previously the day before received fresh caspase inhibitor media as treatment. PAMP transfection was performed 2 h post zVAD treatment. Cells were harvested or assayed as appropriate either by immunoblot or incucyte.
Statistical analysis
Statistical analyses were carried out as indicated in the figure legends. Statistical analyses were performed in GraphPad Prism.
Results
RAR induces parallel cell intrinsic innate immune activation and cell death signaling in HCC cell lines
We manufactured RAR and control XRNA. The XRNA is a 100 nt RNA encoding the HCV genome 3’ nontranslated region (3’ NTR) harboring 3 stem loops but lacking the poly-uridine motif. Both RAR and XRNA are single-stranded (ss) RNA and contain 5’ppp. XRNA does not activate RIG-I and therefore serves as an HCV genome-derived and length-matched nonagonist ssRNA control to RAR (Saito et al., 2008; Schnell et al., 2012; Stone et al., 2013). Both ssRNAs were produced through a large-scale T7 RNA polymerase production system programmed by template DNA and purified as previously described (Saito et al., 2008; Schnell et al., 2012; Stone et al., 2013). To confirm that the manufactured lot of RAR has biological function as a RIG-I agonist in the different HCC lines, we transfected each line with RAR or XRNA control and analyzed via immunoblot assay for innate immune gene/protein expression (Kell et al., 2015; Saito et al., 2008). Huh7 and HepG2 cells were transfected with RAR or XRNA each at a concentration of 250 ng/mL or were mock-transfected with transfection reagent (TFX) treatment only. Protein extracts were collected at 8, 16, and 24 h post-transfection and were subject to immunoblot assay to measure the abundance of RIG-I, MAVS, IRF3, phosphoserine (p)-IRF3, IFIT1, and Actin (internal gel-loading control). As expected, RAR-induced cell intrinsic innate immune activation and response as marked by the expression of RIG-I, P-IRF3, and IFITI over the time course, but protein expression was not induced by either XRNA or mock-transfection (Fig. 1A and B).
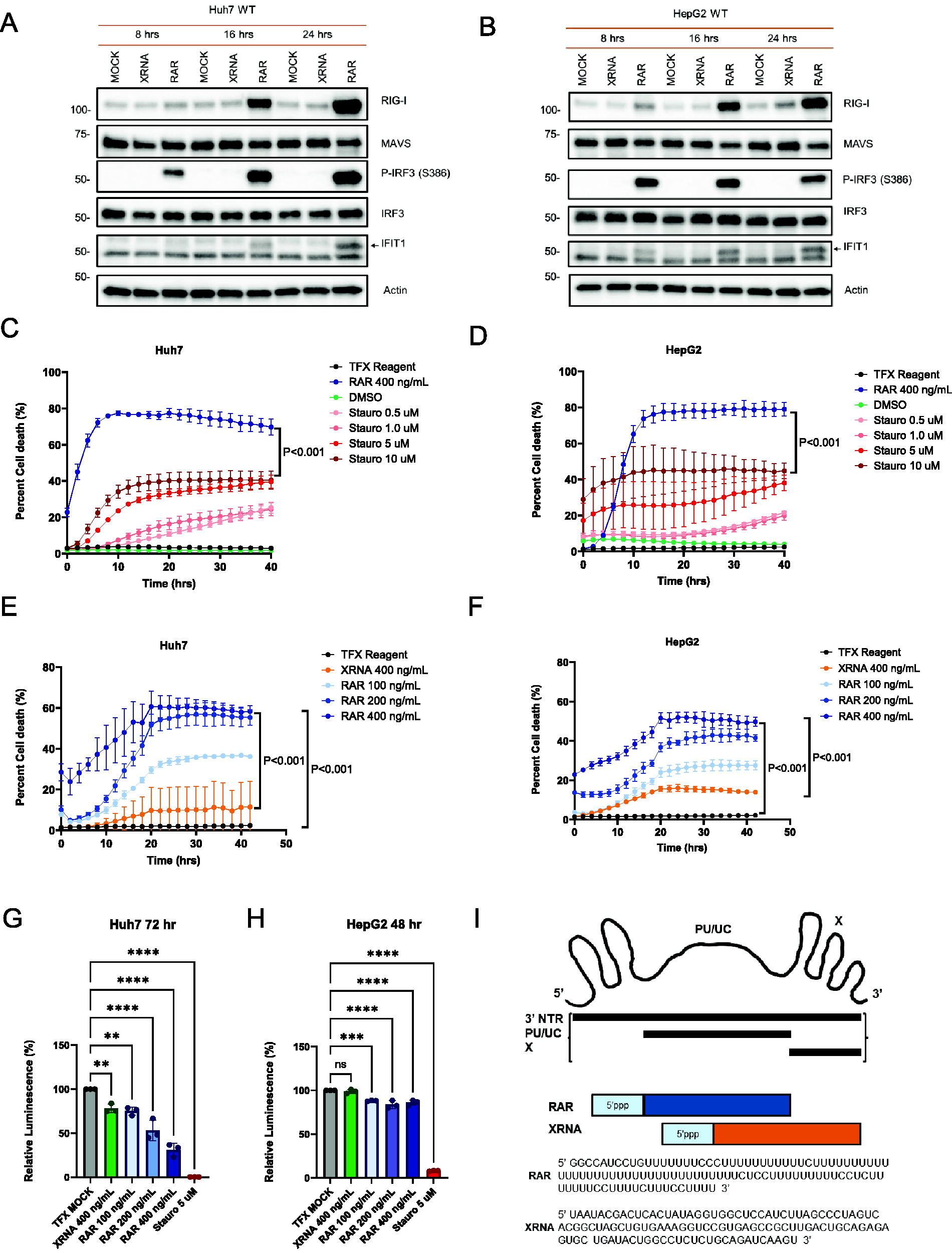
RAR induces cell death in hepatocellular carcinoma cells. Immunoblot analyses showing that RAR is a RIG-I agonist that induces cell intrinsic innate immune gene expression in Huh7
A fluorescence microscopy real-time cell death assay measuring cell uptake of Sytox green, a cell-permeable dye that penetrates compromised membranes characteristic of dead cells (Davis and Gale, 2023), was utilized to assess cell death signaling potential of RAR. Staurosporine (Stauro) treatment of cells was used as a positive control inducer of cell death for treatment of Huh7 and HepG2 cells (Belmokhtar et al., 2001; Malsy et al., 2019). Both Huh7 and HepG2 cells were sensitive to increasing concentrations of staurosporine over a 48-h monitoring period to establish our cell death assay. Cultures of each cell line were transfected in parallel with RAR or XRNA, mock transfected with TFX, or treated with vehicle (DMSO), and monitored for Sytox green uptake. While XRNA control neither induced innate immune activation nor cell death, high dose (400 ng/mL) RAR was found to be a strong inducer of cell death when compared directly to staurosporine treatment (Fig. 1C and D), and each cell line displayed a RAR-induced cell death dose–response (Fig. 1E and F).
To confirm that RAR is inducing cell death, we applied a cell viability assay to measure death of Huh7 and HepG2 cells 72 h after RAR treatment/transfection. Huh7 and HepG2 cells were mock-transfected (TFX), transfected with negative control XRNA (400 ng/mL), treated with positive cell death control Stuarosporine (5 μM), or transfected with RAR (100, 200, and 400 ng/mL). Cell viability was measured as luminescence, 72 h after transfection, using Cell Titer-Glo 2.0 reagent. The Cell Titer-Glo 2.0 reagent determines the number of viable cells in culture by quantifying ATP, which marks metabolically active cells. Viability was quantified as luminescence readout, which is directly proportional to the number of viable cells. As expected, Mock and XRNA had the highest levels of viability. Staurosporine had a significant reduction in viability, confirming it as potent inducer of cell death. Importantly, viability decreased in a dose-dependent manner following RAR treatment/transfection, supporting the previous findings that RAR induces cell death in a dose-dependent manner. Thus, RAR is a potent agonist that drives cell intrinsic innate immune activation in Huh7 and HepG2 cells and directs parallel cell death signaling in these HCC cell lines. Figure 1I shows a schematic of RAR and XRNA structure.
Cell intrinsic innate immune and cell death signaling by RAR are RIG-I-dependent and propagated through the RLR pathway
To assess RAR signaling specificity in cell death, we utilized a series of CRISPR knockout (KO) cell lines lacking expression of RIG-I, MDA5, MAVS, IRF3, or type I IFN receptor (IFNAR), as well as HepG2 cells lacking RIG-I, or MAVS, while cells expressing a nontargeting guide RNA (NTC) were used as controls (Fig. 2A and B). Compared to parental (wild type) or NTC Huh7 cells, HCC KO cells lacking RIG-I, MAVS, or IRF3 fail to induce p-IRF3 or increased expression of RIG-I or IFIT1 in response to RAR treatment. Huh7-IFNAR KO cells displayed reduced p-IRF3 accumulation following RAR treatment (Fig. 2A). Real-time reverse transcription quantitative PCR assay demonstrated that RAR, but not XRNA, treatment of Huh7-NTC cells induces mRNA expression of a range of RIG-I responsive genes including IFITM1, IFIT1, MX1, IFN-β, IL-6, and OAS1 (Fig. 2C). In contrast, Huh7 KO cells lacking RIG-I, MAVS, or IRF3 were recalcitrant to RAR-mediated gene upregulation (Fig. 2C). Importantly, RAR treatment (400 ng/mL) of Huh7 parental/wild-type cells robustly induced cell death but cells lacking RLR pathway components RIG-I, MAVS, or IRF3 were refractory to RAR- induced cell death and remained viable through the 50-h observation period (Fig. 2D). Similarly, RAR treatment resulted in cell death of HepG2-NTC cells but minimal death in HepG2 KO cells lacking RIG-I or MAVS (Fig. 2E). Thus, RIG-I-mediated RLR signaling is essential for RAR-induced death of Huh7 and HepG2 HCC cells, and cell death signaling occurs in parallel with RIG-I-dependent RLR signaling following RAR treatment.
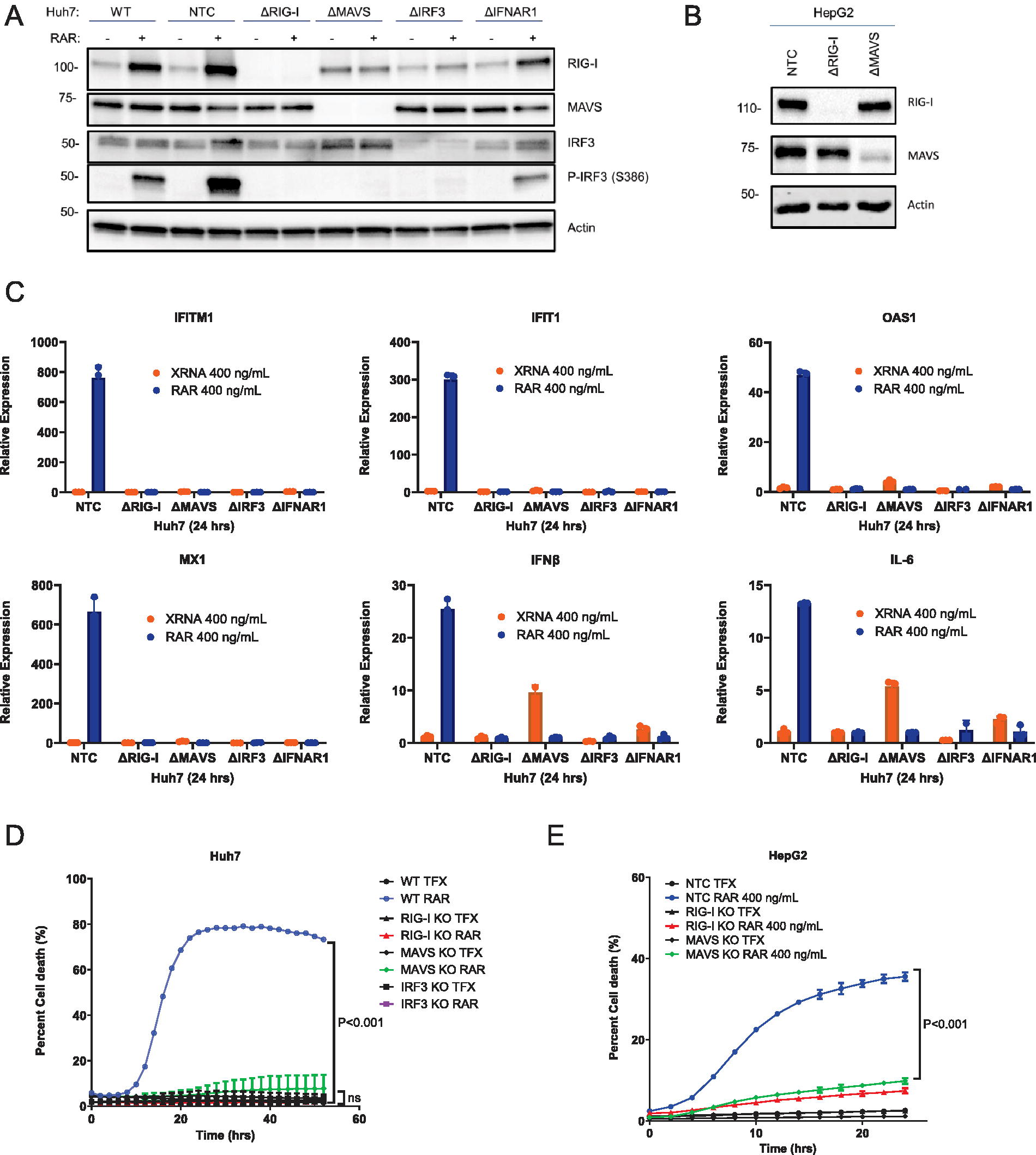
RAR-induced cell death is dependent on RIG-I and RLR pathway signaling. Huh7
Type I interferon potentiates RAR-induced cell death signaling
RIG-I and RLR signaling directs IFN expression and mediates an IFN-feedback loop in which IRF3-target genes and interferon-stimulated genes (ISGs) together function to enhance and diversify the innate immune response (Chow et al., 2018; Loo and Gale, 2011). We therefore assessed the impact of IFN-β treatment on RAR-induced cell death signaling. Huh7 cells induce ISG expression (MX1) in response to IFN-β treatment (Fig. 3A). When treated with IFN-β alone, parental/wild-type Huh7 (Fig. 3B) or HepG2 (Fig. 3C) cells do not undergo cell death even at high treatment concentration, but parallel treatment with RAR results in cell death of each within the observation period. We note that RAR treatment induces a level of p-IRF3 and IRF3-target gene expression independent of IFN signaling (Sumpter et al., 2005; Yoneyama et al., 2005, 2004) as observed in Huh7-IFNAR KO cells (see Fig. 2A). We therefore interrogated if IFN signaling amplifies RAR-induced cell death. We conducted a cell death assay comparing Huh7 parental/wild-type cells to Huh7-IFNAR1 KO cells that were mock-treated or treated with RAR. Compared to Huh7 parental/wild-type cells, IFNAR KO cells display an approximate 50% reduction in cell death over the observation period, suggesting that IFN signaling potentiates RAR-induced cell death (Fig. 3D). To determine if IFN-β treatment can indeed potentiate RAR-induced cell death, Huh7 parental/wild-type cells were pretreated with 100 units (U) of IFN-β or culture media alone for 24 h followed by RAR treatment, and cell death was monitored over a subsequent 36-h observation period. IFN-β pretreatment followed by RAR resulted in response with enhanced kinetics and frequency of cell death compared to RAR treatment alone (Fig. 3E). We also confirmed that IFN-β alone was not sufficient to induce cell death, showing that cell death signaling is RAR dependent and is potentiated by IFN signaling.

Type I interferon potentiates RAR-induced cell death in HCC cells.
RAR induces caspase-dependent death in HCC cells
RIG-I signaling and IRF3 are linked to caspase activation and apoptosis induced by virus infection and other stimuli (Lee and Kim, 2007). To determine if RAR signaling of cell death links with signaling of caspase activation, we conducted immunoblot assay to evaluate the abundance of caspase-3 and substrate poly (ADP-ribose) polymerase (PARP), which are major markers of caspase-mediated cell death signaling (Huang et al., 2022). Parental/wild-type Huh7 (Fig. 4A) and HepG2 (Fig. 4B) cells that were treated with RAR in the presence or absence of the pan-caspase inhibitor zVAD-FMK (zVAD) (Kim et al., 2005) displayed RAR-induced p-IRF3 and IFIT1 expression. In the absence of zVAD treatment cleaved caspase-3 and cleaved PARP accumulated in RAR-treated cells concomitant with p-IRF3 and IFIT1, but zVAD treatment prevented the accumulation of cleaved caspase-3 and PARP (Fig. 4A and B). We also found that zVAD treatment of cells suppressed RAR-induced cell death (see Fig. 4C and D). These observations indicate that RAR directs caspase-dependent cell death signaling in HCC cells. Together, our observations demonstrate that RAR can direct apoptotic cell death in HCC cell lines concomitant with innate immune activation and that IFN signaling serves to potentiate cell death signaling.

RAR induces caspase-dependent death of HCC cells. Western blots of Huh7 cells
Discussion
Our study shows that RAR induces caspase-dependent cell death in hepatic tumor cell lines, that RIG-I and RLR signaling are required for caspase activation of this cell death program, and that type I IFN potentiates RAR-mediated cell death signaling. Based on previously published work (Chattopadhyay et al., 2016, 2010; Chow et al., 2018; Kell et al., 2015; Loo and Gale, 2011; Saito et al., 2008; Schnell et al., 2012; Stone et al., 2013; Sumpter et al., 2005; Yoneyama et al., 2005) and the current study, we propose a model for RAR-induced cell death (Fig. 5) as follows: (1) RAR triggers RIG-I activation and the downstream RLR signaling pathway following its binding by RIG-I (Schnell et al., 2012). RIG-I then signals downstream through MAVS and IRF3 to induce gene expression including IRF3 target genes. (2) IFN production from RLR signaling facilitates an IFN-signaling feedback loop that drives ISG expression including RIG-I. Because RIG-I is an interferon-stimulated gene, cell death signaling is potentiated by IFN resulting in increased kinetics and frequency of RAR-induced cell death. Importantly, interferon-β treatment alone will not induce cell death without RAR treatment to mediate activation of RIG-I and RLR signaling. (3) Caspase-mediated cell death activation: RAR-induced RIG-I signaling culminates in the activation of caspase-mediated apoptosis characterized by the activation of effector caspases-3 and PARP cleavage in a zVAD-sensitive manner.
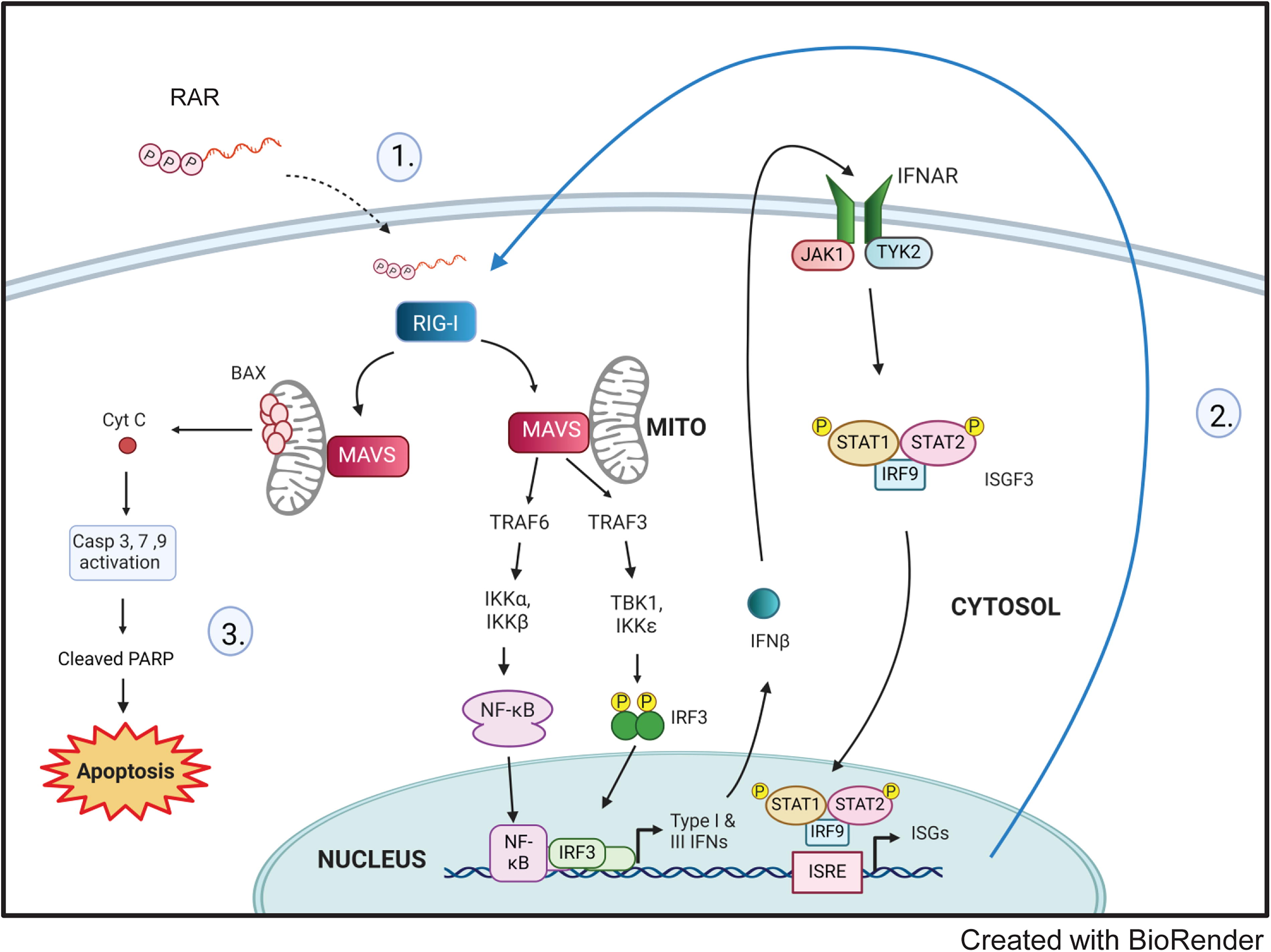
Model of RAR induced RIG-I-mediated caspase-dependent death of HCC cells.
RIG-I is a cytoplasmic PRR ubiquitously expressed in human tissues, and its role extends beyond that of a pattern recognition receptor (Iurescia et al., 2020; Loo and Gale, 2011; Saito et al., 2008). In cancer cells, the two major outcomes of RIG-I signaling are the production of interferons and the activation of programmed tumor apoptosis (Besch et al., 2009; Duewell et al., 2014; Glas et al., 2013; Iurescia et al., 2020; Kaneda, 2013; Koren and Fuchs, 2021). Host defense responses against virus-infected cells and against tumors share intrinsic molecular features, it is therefore theorized that RIG-I activation is selective in cancer cells as a way to mimic a viral infection and either induce immunogenic cell death or direct cancer cell apoptosis (Elion et al., 2018; Iurescia et al., 2020; Kaneda, 2013; Koren and Fuchs, 2021). Interferons are pleiotropic immune-modulatory cytokines that are well known for their essential role in host defense responses against viruses, bacteria, and other pathogenic microorganisms (Besch et al., 2009; Cheon et al., 2014; Chow et al., 2018; Kotredes and Gamero, 2013; Loo and Gale, 2011; Stolzer et al., 2021; Su et al., 2007). Interferon signaling results in the induction of IFN-stimulated genes influencing different cellular pathways including direct antiviral responses, immune modulation, or cell death (Kell and Gale, 2015; Schnell et al., 2012; Stolzer et al., 2021). Interferons can induce or modulate gene expression of essential signaling players of cell death pathways including apoptotic, pyroptotic, and necroptotic cell death. In our study, we show that RAR, an RLR agonist, induces caspase-dependent cell death and that this cell death program can be modulated by interferon. While interferon alone does not induce cell death in our HCC model, interferon enhances the RAR-induced RLR-mediated cell death program. IRF3 was found to be another key regulator of RAR-induced RLR-mediated cell death. IRF3 also plays a key role in several other forms of cell death, including transcription-dependent and transcription-independent cell death pathways (Chattopadhyay et al., 2016, 2010; Chattopadhyay and Sen, 2017). For example, the RIG-I induced pathway of apoptosis, known as RIPA, utilizes transcription-incompetent IRF3 mutants in a manner to direct transcription-independent apoptotic cell death (Chattopadhyay et al., 2016; Chattopadhyay and Sen, 2017). Ongoing studies will define the signaling processes by which RAR promotes caspase-dependent cell death.
RLR agonists can trigger tumor-resident innate immune receptors to initiate apoptotic programs in tumor cells, making RAR and other RIG-I ligands attractive targets for chemotherapeutic development (Chattopadhyay and Sen, 2014). Double-stranded (ds) RNA species have been studied for their ability to induce various forms of cell death in different cancer cell models. Moreover, virally derived double-stranded RNA or synthetic analogs have been shown to activate several innate immune receptors, including RIG-I, TLR3, MDA5, and PKR (Chattopadhyay and Sen, 2014). Once stimulated, these factors initiate signaling pathways that lead to the production of proinflammatory cytokines and type I interferon, apoptosis, and translational inhibition (Chattopadhyay and Sen, 2014). Clinical trials for breast and gastric cancer that included dsRNA in combination with standard care showed improved overall survival and progression-free survival (Andre et al., 2004; Elion et al., 2018; Stolzer et al., 2021). In melanoma, breast, prostate, and HCC cells, dsRNA has been shown to trigger a caspase-dependent apoptotic response (Andre et al., 2004; Bek et al., 2019; Besch et al., 2009; Grützner et al., 2024; Helms et al., 2019; Palchetti et al., 2015; Zhou et al., 2020). RAR is generated from the 3’ UTR region of the HCV genome. HCV is a hepatotropic, positive-sense, single-stranded RNA virus that unless interrupted by antiviral therapy establishes a chronic infection in most people after acute exposure (Kell et al., 2015). Previous studies showed that RIG-I recognition of HCV is dependent on the 100 nt poly-uridine/cytosine-rich motif found in the 3’ nontranslated region (3’ NTR) partially encoded by RAR (Saito et al., 2008; Schnell et al., 2012). Though several other RIG-I agonists have been studied for their activation of caspase-mediated tumor cell death, our current study represents the first examination of RAR in this context. Our observations show that RAR drives caspase-dependent cell death and further reveal that it induces apoptosis via PARP cleavage. These findings pose RAR as a promising agonist to study for oncolytic therapy driven by RLR-activated caspase-dependent cell death.
Conclusions
In summary, our study demonstrates that RAR induces RIG-I and RLR signaling-dependent innate immune activation, caspase activation, and cell death signaling in HCC cells and that RAR induction of cell death is potentiated by type I interferon. RIG-I and RLR signaling is increasingly recognized as important components of tumor control (Besch et al., 2009; Cheon et al., 2014; Elion and Cook, 2018; Glas et al., 2013; Grützner et al., 2024; Sugii et al., 2021). We conclude that RAR treatment could contribute to strategies for the control of liver cancer and other cancer types.
Footnotes
Authors’ Contributions
B.U. conducted experiments, analyzed data, and wrote the paper. I.B. conducted and analyzed experiments. B.B. and M.D. produced RAR and XRNA, conducted and analyzed experiments, and edited the paper. S.R. designed experiments and edited the paper. R.S. designed experiments and edited the paper. M.G. directed the research, designed and analyzed experiments, and edited the paper.
Author Disclosure Statement
M.G., B.B., S.R., and M.D. have financial interest in HDT Bio. B.U., I.B., and R.S. have no conflicts to disclose.
Funding Information
This work was supported by NIH grants AI145296, AI104002, and AI183793 (MG) and NIAID T32AI106667 (MG and BSU).