
Abstract
Alphaviruses (family Togaviridae) are a diverse group of positive-sense RNA (+ssRNA) viruses that are transmitted by arthropods and are the causative agent of several significant human and veterinary diseases. Interferon (IFN)-induced proteins with tetratricopeptide repeats (IFITs) are a family of RNA-binding IFN-stimulated genes (ISGs) that are highly upregulated following viral infection and have been identified as potential restrictors of alphaviruses. The mechanism by which IFIT1 restricts RNA viruses is dependent on self and non-self-discrimination of RNA, and alphaviruses evade this recognition via their 5′ untranslated region (UTR). However, the role of IFIT2 during alphavirus replication and the mechanism of viral replication inhibition is unclear. In this study, we identify IFIT2 as a restriction factor for Venezuelan equine encephalitis virus (VEEV) and show that IFIT2 binds the 3′ 3′UTR of the virus. We investigated the potential role of variability in the 3′UTR of the virus affecting IFIT2 antiviral activity by studying infection with VEEV. Comparison of recombinant VEEV clones containing 3′UTR sequences derived from epizootic and enzootic isolates exhibited differential sensitivity to IFIT2 restriction in vitro infection studies, suggesting that the alphavirus 3′UTR sequence may function in part to evade IFIT2 restriction. In vitro binding assays demonstrate that IFIT2 binds to the VEEV 3′UTR; however, in contrast to previous studies, VEEV restriction did not appear to be dependent on the ability of IFIT2 to inhibit translation of viral RNA, suggesting a novel mechanism of IFIT2 restriction. Our study demonstrates that IFIT2 is a restriction factor for alphaviruses and variability in the 3′UTR of VEEV can modulate viral restriction by IFIT2. Ongoing studies are exploring the biological consequences of IFIT2-VEEV RNA interaction in viral pathogenesis and defining sequence and structural features of RNAs that regulate IFIT2 recognition.
Introduction
Alphaviruses (family Togaviridae) are a diverse group of positive-sense RNA (+ssRNA) viruses that are transmitted by arthropods and are the causative agent of several significant human and veterinary diseases. Venezuelan equine encephalitis virus (VEEV) belongs to the VEE complex of alphaviruses and is responsible for periodic epizootic outbreaks of disease in equines in South, Central, and North America (reviewed in Weaver et al., 2004). The VEE serocomplex consists of numerous different species that can be divided into 6 subtypes (I–VI). VEEV belongs to subtype I and can be further classified as epizootic (IAB and IC) or enzootic (ID and IE). During inter-epizootic periods, VEEV is transmitted via a sylvatic cycle involving Culex spp. mosquitoes and rodents, which serve as reservoirs for the virus. These enzootic subtypes (ID and IE) are considered equine avirulent and not associated with significant morbidity or mortality in these species. In contrast, epizootic variants (IAB and IC) are transmitted by several different species of mosquito and are associated with high titer viremia in equines, which serve as amplification hosts during epizootic outbreaks. Epizootic subtypes are known to arise from mutation of sympatric enzootic viruses, and the epizootic phenotype has been shown to correlate with type I interferon (IFN) resistance (Spotts et al., 1998). Genetic studies have previously mapped the key epizootic determinants to the E2 glycoprotein, which serves as the viral attachment protein (Bernard et al., 2000; Brault et al., 2002a; Brault et al., 2002b; Kinney et al., 1993). Mutations in E2 sequences of epizootic subtypes have been shown to support higher levels of replication in horses and greater virulence, as well as adaptation to epizootic mosquito vectors (Greene et al., 2005b; Brault et al., 2002b). Although these mutations alone have been shown to be sufficient to confer epizootic phenotypes, epizootic subtypes also acquire mutations outside of this region, suggesting that additional genetic changes may contribute to the emergence of epizootic VEEV (Greene et al., 2005a). None of the studies has investigated the effect the sequence diversity in the 3′ untranslated region (3′UTR) of VEEV has on viral pathogenesis.
Alphaviruses are sensitive to type I IFN, and IFN-stimulated genes (ISGs) are known to be the effectors of alphavirus restriction. We have previously identified IFIT1 as a restriction factor for VEEV and other alphaviruses (Hyde et al., 2014). While IFIT1 recognizes non-self-viral RNA by preferentially binding to and restricting 7-methylguanisine (m7G) capped viral RNA (cap 0), alphaviruses can evade IFIT1 by encoding stable RNA structures within their 5′UTR (Hyde et al., 2014; Habjan et al., 2013; Szretter et al., 2012; Kimura et al., 2013). However, the role of IFIT2 in alphavirus replication and pathogenesis has not yet been defined. Like IFIT1, IFIT2 also binds RNA (Yang et al., 2012) and has been shown to protect against lethal neuropathogenesis induced by diverse RNA viruses including flaviviruses, coronaviruses, and rhabdoviruses (Fensterl et al., 2012; Cho et al., 2013; Butchi et al., 2014; Fensterl et al., 2014; Wetzel et al., 2014; Davis et al., 2017; Saha et al., 2006; Sharma et al., 2023; Poddar et al., 2024). Despite this, the precise mechanisms by which IFIT2 inhibits replication and pathogenesis of such diverse viruses are still not fully understood. Furthermore, IFIT2 has been implicated in diverse cellular processes, including RNA binding (Yang et al., 2012; Fleith et al., 2018), translational regulation (Terenzi et al., 2005; Berchtold et al., 2008), cell death (Stawowczyk et al., 2011; Feng et al., 2014; Wang et al., 2016; Chen et al., 2017; Zhang et al., 2017), cancer development (Lai et al., 2013; Lai et al., 2016; Wang et al., 2016; Jia et al., 2017; Chen et al., 2018; Shen et al., 2018), and immune signaling (Siegfried et al., 2013; Butchi et al., 2014; Imaizumi et al., 2014; Imaizumi et al., 2016). Although IFIT2 has predominantly been demonstrated to function as a viral restriction factor in vitro and in vivo, more recent studies suggest that IFIT2 may promote viral mRNA translation and replication (Tran et al., 2020), suggesting that IFIT2 may function in a proviral manner in different contexts.
Here, we identify IFIT2 as a restriction factor for VEEV using an shRNA-based screening approach and show that IFIT2 also protects mice from VEEV pathogenesis in vivo. We show that IFIT2 binds AU-rich RNA located in the 3′ end of the viral genome and that changes in the 3′UTR sequence alter the sensitivity of VEEV to IFIT2-dependent inhibition in a cell type-dependent manner. We further show that replication of viruses encoding 3′UTR sequences from epizootic and enzootic VEEV subtypes is differentially affected in an IFIT2-dependent and IFIT2-independent manner, suggesting that IFIT2 recognition of viral RNA is likely modulated by RNA structure. Last, we show that the mechanism by which IFIT2 inhibits VEEV is independent of viral RNA translation and suggest possible mechanisms by which IFIT2 restricts alphavirus replication.
Materials and Methods
Lentiviral transduction and flow cytometry
HeLa cells were transduced with lentiviruses encoding shRNA. The bicistronic vector (pGIPZ) co-expresses the shRNA and GFP, which are driven by IRES and cytomegalovirus immediate early promoters, respectively. Individual shRNA constructs [control (NSC, catalog # RHS4346) and human STAT2 and human IFIT1 (Supplementary Table S1)] were packaged into lentiviral vectors following the manufacturer’s instruction (Open Biosystems). Two days post-transduction, cells were treated with 10 IU/mL of human IFN-β (PBL interferon source) for 6 h and then infected with TC83 at an multiplicity of infection (MOI) of 1. Twenty-four hours post-infection, cells were trypsinized, washed once in Hanks balanced salt solution (HBSS), fixed in 1% paraformaldehyde for 10 min at room temperature, and then permeabilized in permeabilization buffer (HBSS, 10 mM HEPES pH 7.3, and 0.1% saponin). Cells were subsequently with incubated with antibodies specific for VEEV (Roehrig et al., 1982) in permeabilization buffer at room temperature for 20 min. After 3 washes, cells were incubated with species-specific Alexa Fluor 647-conjugated secondary antibodies (Molecular Probes) in permeabilization buffer. Cells were analyzed for GFP expression (shRNA expression) and viral antigen by flow cytometry on a BD FACS Array flow cytometer (BD Biosciences). Data were processed using FlowJo analysis software (Tree Star, Inc.). The initial preliminary shRNA screen was performed in duplicate 2 times independently, and the fold increase in VEEV-positive cells was calculated by comparing data to a nonsilencing control shRNA, and z-scores calculated. Based on these data, a subset of genes was chosen for further validation based on the following criteria: (1) hits with a z-score >2, (2) hits for which more than 2 shRNAs against the same gene exhibited 2-fold or greater VEEV-positive cells relative to shNSC; and (3) the validation screen was performed as described above using 10 IU/mL of IFN-β stimulation and was performed in quadruplicate 4 times independently.
Antibodies and cell lines
Vero C1008 and BHK cells were obtained from American Type Culture Collection (ATCC). All cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% defined heat-inactivated FBS (HyClone),
Generation of primary macrophages
Primary 3-day bone marrow-derived macrophages (BMDM) were generated as previously described (Hyde et al., 2014). BMDM were grown in DMEM supplemented with 10% FBS, 200 mM L-glut, 10,000 U/mL penicillin (Sigma), 10 mg/mL streptomycin (Sigma), and 20% L929-conditioned cell supernatant. To harvest cells, bone marrow from femurs and tibias from the indicated mouse strains was plated for 1 h (37°C and 5% CO2) in DMEM medium without supplements to allow attachment of macrophages. The nonadherent dendritic cells were removed and cultured separately in appropriate medium. Adherent macrophages were then cultured in appropriate media. After 6 days, nonadherent adherent macrophages were scraped off, counted, and seeded for experiments.
Generation of Raw264.7 Ifit2−/− CRISPR cells
A doxycycline-inducible CRISPR/Cas9 expression vector (pSBtet-puro-Cas9-U6) was generated by cloning the Cas9-U6 portion of pX459 [Addgene #62988; (Ran et al., 2013)] into pSBtet-pur [Addgene #60507; (Kowarz et al., 2015)]. Cas9 was first cloned into pSBtet-pur using the following primers: Cas9.F: 5′-CATGAGACCGGTGCCACCATG-3′, Cas9.R: 5′-CATGAGGCGGCCGCCTACTTTTTCTTTTTTGCCTGGCCG, pSBtet-pur.F: 5′-CATGAG GCGGCCGCCTTCC-3′, pSBtet-pur.R: 5′-CATGAGACCGGTGGTGGCCGATATCTCAGAG. Cas9 was ligated into the pSBtet-pur backbone using 5′ AgeI and 3′ NotI restriction sites. The U6 promoter was then cloned into this new plasmid using the following primers: U6.F 5′-ACTACAGGTACC GAGGG-3′, U6.R 5′-TCAGTCCTAGGTCTAGAGC-3′, pSBtet-pur-Cas9.F 5′-TCAGTCCTAGGTCTAGAGC-3′, pSBtet-pur-Cas9.R 5′-ATGAAGGTACCACATTTGTAGAGGTTTTACTTGC-3′. U6 was ligated into pSBtet-pur-Cas9 using 5′ KpnI and 3′ AvrII restriction sites. As the new pSBtet-pur-Cas9-U6 plasmid contained an additional BbsI site, this was removed using site-directed mutagenesis and the following primers: dBbsI.F 5′-TTGG GAAGAT AATAGCAG-3′, dBbsI.R 5′-CTGCTATTATCTTCCCAA-3′.
Sequence-specific gRNA sequences were designed using the Broad Institute Genetic Perturbation Platform gRNA design tool to target mouse Ifit2 (accession # NM_008332). The following primers were used to generate Ifit2 gRNA oligonucleotides, which were cloned into pSBtet-puro-Cas9-U6 as described previously (Ran et al., 2013): Ifit2.g13.F: 5′-CACCGCACTGCAGAGGTCTAAATG-3′, Ifit2.g13.R: 5′-AAACCATTTAGACCTCTGCAGTGC-3′; Ifit2.g25.F: 5′-CACCGATCAGAAGTCTGGTCACCTG-3′, Ifit2.g25.R: 5′-AAACCAGGTGACCAGACTTCTGATC-3′; Ifit2.g38.F: 5′-CACCGCAGGATTCTCAATCCTGTAG-3′, Ifit2.g38.R: 5′-AAACCTACAGGATTGAGAATCCTGC-3′. Raw264.7 Ifit2 CRISPR cells were generated by electroporating low passage Raw264.7 cells with Ifit2 pSBtet-puro-Cas9-U6 using Amaxa Nucleofector II and Amaxa Cell Line Nucleofector Kit V (Lonza). Cells were selected with puromycin 3 days post-nucleofection, and Cas9/gRNA expression induced at 7 days post-nucelofection. Cells were treated for 7–14 days with doxycycline and knock out (KO) efficiency of bulk cells validated using western blotting and detection with mouse anti-ISG54 (6.H9).
Generation of full-length and recombinant viruses
Construction of the full length TC83 VEEV infectious clone has been described (Kinney et al., 1993). The following VEEV epizootic and enzootic 3′UTR sequences were introduced into the TC83 infectious clone: (IC) TGAACATAGCAGCAATTGGCAAGCTGCTTATATAGAACTTGCGGCGATTGGCATGCCGCTTTAAAATTTTATTTTATTTTCTTTTCTTTTCCGAATCGGATTTTGTTTTTAATATTTC; (ID) TGAACATAGCAGCAATTGGCAAGCTGCTTATATAGAACTCGCGGCGATTGGCATGCCGCTTTAAAATTTTATTTTATTTTCTTTTCTTTTCCGAATCGGATTTTGTTTTTAATATTTC; (IE) TAGAATTAGCAGCGATTGGCATGCTGCTTGTAAAGTTTTATTACAAATAACGTGCGGCAATTGGCGAGCCGCTTTAATTAGAATTTTATTTTCTTTTACCATAATCGGATTTTGTTTTTAATATTTC. Point mutations were introduced by site-directed mutagenesis using Phusion High-Fidelity PCR kit (NEB) using primers listed in Supplementary Table S2.
Plasmids were linearized at MluI restriction sites located downstream of the poly(A) tail, and genomic RNA was transcribed from the SP6 promoter in the presence of N7mG cap analog using the SP6 mMessage mMachine kit (Ambion); 1 × 107 BHK21 cells were electroporated with ∼2 µg of in vitro transcribed RNA using a GenePulser Xcell electroporator (Bio-Rad) to generate P0 virus stocks.
Focus-forming assays
Focus-forming assays (FFAs) were performed in 96-well plates by adapting an assay developed for flaviviruses (Fuchs et al., 2011). Vero E6 monolayers were infected with serial 10-fold dilutions of infectious samples for 1 hour at 37°C, then overlaid with 100 µL per well of medium (0.5x DMEM, 5% FBS) containing 1% carboxymethylcellulose, and incubated for 20–22 h at 37°C with 5% CO2. Cells were then fixed by adding 100 µL per well of 2% paraformaldehyde directly onto the overlay at room temperature for 2 h. After removal of overlay media and fixative, cells were washed 3× with phosphate-buffered saline (PBS) and incubated with antibodies specific for VEEV E2 glycoprotein (gift of Dr. Michael Diamond; see above) for 2 h at room temperature in FFA permeabilization buffer [1× PBS, 0.1% saponin, and 0.1% bovine serum albumin (BSA)]. Cells were washed 3× in ELISA wash buffer (1× PBS, 0.05% triton X-100), then incubated with species-specific horse radish peroxidase (HRP)-conjugated secondary antibodies (Sigma and ThermoFisher) for 1 h at room temperature in FFA permeabilization buffer. Monolayers were washed 3× with ELISA buffer and foci were developed by incubating in 50 µL/well of TrueBlue peroxidase substrate (KPL) for 5–10 min at room temperature, after which time cells were washed twice in water. Well images were captured using Immuno Capture software (Cell Technology Ltd.), and foci counted using BioSpot software (Cell Technology Ltd.). All samples were titered in duplicate and calculated titers averaged for each duplicate.
Mouse experiments
All mouse experiments were performed in compliance and with the approval of the Washington University School of Medicine and University of Washington Animal Studies Committees. C57BL/6 wild type (WT) mice were obtained from Jackson laboratories, and congenic Ifit2 −/− mice were generated previously (Daffis et al., 2010; Fensterl et al., 2012) (gift of Dr. Ganes Sen) (Szretter et al., 2012). Sixteen-week-old male and female mice were used in all in vivo infection experiments. For subcutaneous infection, 102 focus-forming units (FFU) of ZPC738 was diluted in PBS in a total volume of 50 µL and delivered via injection into the rear footpad.
Viral growth kinetic assays
Murine embryonic fibroblasts (MEF) and Raw264.7 cells were maintained in DMEM supplemented with 10% defined heat-inactivated FBS,
Translation assays
Construction of the VEEV 3′UTR translation reporters was similar to that described previously (Tesfay et al., 2008; Hyde et al., 2014). Mutations derived from epizootic and enzootic 3′UTR sequences were introduced by restriction digestion and ligation. The following primers were used to amplify 3′UTR sequences from infectious clones (described above): VEEV F: TAGTTAATTAAGATCAGCCGTAATTATTATAATTGG VEEV R: GCGGCCGCTCAAGAATTAATTCCCctcgaC. Fragments were amplified using Q5 high fidelity polymerase (NEB), purified using AMPure XP beads (Beckman Coulter), digested with PacI and NotI (NEB), and ligated into the VRLF vector, which was similarly digested. Reporter RNAs were in vitro transcribed and co-transcriptionally capped using the mMESSAGE mMACHINE T7 kit (Ambion) according to the manufacturer’s instructions, purified using RNeasy Mini Kit (Qaigen). WT and Ifit2 −/− MEF were treated with 0 or 10 IU/mL of IFN-β (PBL Interferon Source) for 6 h. Reporter assays were performed as previously described (Gardner et al., 2008; Hyde et al., 2014) using 2 μg (MEF) or 10 µg (Raw264.7) of RNA for each electroporation reaction, and cells were plated in 96-well plates. Cells were collected at indicated time points by centrifugation at 1,500g for 3 min, washed 1× in DPBS (Gibco), and lysed in 1× cell culture lysis buffer (Promega). Firefly luciferase assays were performed on 20 µL of cell lysates using Luciferase Assay System (Promega) according to the manufacturer’s instructions. Luciferase activity was measured on a BioTek Synergy microplate luminometer. Relative light unit values were normalized to protein concentration using a standard BCA Protein assay (Pierce).
Generation of mouse recombinant IFIT2 expression vector
An expression vector encoding human IFIT2 (hIFIT2) [pET28a(+)-6xHis-TEV-IFIT2, Addgene; (Katibah et al., 2013)] was modified to express mouse IFIT2 (mIFIT2). A synthetic gene fragment encoding mIFIT2 (accession number NM_008332) flanked by NheI (5′) and XhoI (3′) restriction sites was synthesized (IDT). The hIFIT2 sequence was removed from pET28a(+)-6xHis-TEV-IFIT2 by digestion with NheI and XhoI, and mIFIT2 ligated into the corresponding sites, to generate the new pET28a(+)-6xHis-TEV-mIFIT2 expression plasmid. Plasmid was transformed into 10-beta cells (NEB) and clone sequence verified by Sanger sequencing.
Recombinant protein expression and purification
pET28a(+)-6xHis-TEV-mIFIT2 plasmid encoding mouse IFIT2 was transformed into Rosetta (DE3) pLysS chemically competent cells. Stationary phase cultures of the resulting transformed bacteria were used to inoculate 1.5 L of LB broth at a 1:100 dilution. Bacterial cultures were grown to OD600 0.4 at 37°C after which protein expression was induced by the addition of 0.5 mM isopropyl β‐D‐1‐thiogalactopyranoside for 20 h at 16°C. Bacteria were harvested by centrifugation at 7,000g for 30 min, and the cell pellets were resuspended in 30 mL of buffer A (20 mM HEPES, pH 7.3, 1.5M NaCl, 2 mM MgCl2, 25 mM imidazole, 10% glycerol, supplemented with 5 mM BME, 1 mM PMSF, and 0.2 mg/mL lysozyme). Resuspended cells were incubated on ice for 20 min, lysed by sonication, and then cleared by centrifugation at 40,000g for 30 min at 4°C. Clarified lysate was bound to 0.5 mL of HisPur Ni-NTA resin (Thermo Scientific). The resin was then washed with 100 mL of buffer A and bound proteins were eluted in buffer B (20 mM HEPES, pH 7.3, 150 mM NaCl, 2 mM MgCl2, 300 mM imidazole, and 10% glycerol supplemented with 5 mM BME). The purity of each fraction was determined by SDS-PAGE analysis followed by Coomassie staining. Fractions containing IFIT2 protein were pooled, supplemented with 0.4 mg mL−1 TEV protease, and dialyzed overnight at 4°C in buffer C (20 mM HEPES, pH 7.3, 150 mM NaCl, 2 mM MgCl2, 10% glycerol supplemented with 1 mM BME). The sample was then passed through 0.5 mL of Ni-NTA resin to remove TEV protease, hexahistidine peptide, and uncut IFIT2. The resulting flow through fraction was analyzed for purity by SDS-PAGE analysis followed by Coomassie staining. The sample was then concentrated, snap frozen, and stored at −80°C until use.
Differential Radial Capillary Action of Ligand Assays
For Differential Radial Capillary Action of Ligand Assays (DRaCALA) comparing binding of rIFIT2 and BSA to the 3′UTR, the 3′ 200 nucleotides of VEEV TC83 were amplified using primers listed in Supplementary Table S2 and using the infectious clone cDNA for TC83. For DRaCALAs comparing binding of rIFIT2 with viral and synthetic RNAs with different AU content, 101–106 nucleotide fragments were amplified using the primers listed in Supplementary Table S3 using the TC83 infectious clone as template. Forward primers contained the sequence for the bacteriophage class III T7 promoter sequences. PCR products were purified and in vitro transcribed using T7 HiScribe kit (NEB). Transcribed RNA products were run on a 5% acrylamide TBE-urea gel and visualized with 0.02% methylene blue in 1× TBE. RNAs were excised from gels and transferred into a 0.6 mL microfuge tube with a hole in the bottom (made by a sterile 22.5G needle). The 0.6 mL tube was placed in a 2 mL microfuge tube and centrifuged at 10,000g for 5 min to crush gel pieces. RNA was eluted by adding 3× volume excess of RNA elution buffer [10 mM Tris–HCL (pH 7.5), 1 mM EDTA, 300 mM NaCl, 0.1% SDS]. Samples were then frozen at −80°C for 15 min and then heated to 95°C for 5 min and placed on a rotator at room temperature overnight. Gel fragments were removed by centrifuging samples though a cellulose acetate filter (Thermo Scientific) and at 10,000g for 3 min at room temperature. RNA was then purified by acid phenol:chloroform extraction. In vitro transcribed RNA (5′-ppp) was dephosphorylated and 5′ end labeled with P32 as follows: 25 pmol of RNA was dephosphorylated using 50U of Antarctic phosphatase (NEB) according to the manufacturer’s protocol. Dephosphorylated RNA was then 5′ end-labeled with 20U T4 PNK (NEB) and 30µCi [g32-P]ATP (Perkin-Elmer). Reactions were incubated for 1.5 h at 37°C. Unincorporated nucleotides were removed using Micro Bio-Spin 6 columns (Bio-Rad) according to the manufacturer’s protocol, and samples were then purified using Mag-Bind Total Pure NGS beads (Omega Bio-Tek). Prior to performing binding assays, the RNA was folded by heating at 95°C for 2 min and slow cooled at room temperature. Binding reactions were performed as follows: 100 fmol of RNA was combined with serial 2-fold dilutions of rIFIT2 and incubated at 37°C for 15 min in binding buffer (50 mM HEPES, 100 mM KCl) supplemented with 5 mM MgCl2, 5% glycerol, 500 ng of yeast tRNA, and 1 mM DTT. One microliter of each reaction was spotted in duplicate onto a 0.45 µM nitrocellulose membrane (Amersham) and allowed to dry at room temperature for 20 min. RNA was then visualized by exposing the nitrocellulose to a phosphor screen, and the resulting signal was measured on a sapphire imager (Azure Biosystems).
Results
An shRNA screen identifies IFIT2 as a restriction factor for Venezuelan equine encephalitis virus
Previously, we identified IFIT1 as a restriction factor for replication of VEEV (Hyde et al., 2014). To identify other ISGs that restrict VEEV replication, we used an shRNA-based approach to knock down expression of ∼250 IFN- and virus-induced genes (Supplementary Table S4; Supplementary Table S5). HeLa cells were transduced with lentivirus expressing a bicistronic reporter consisting of GFP and shRNA-targeting individual host genes or a nonsilencing control shRNA (shNSC). Forty-eight hours post-transduction, cells were stimulated with human IFN-β to activate ISG expression and then infected with VEEV TC83 (MOI 1). At 24 h post-infection (hpi), cells were fixed and analyzed by flow cytometry to quantify the number of VEEV-positive cells, as determined by staining for VEEV E2 protein. Knockdown of VEEV restriction factors was anticipated to lead to an increase in the number of VEEV E2-positive cells of the transduced population (shRNA-expressing; GFP positive). As expected, we observed that knockdown of genes involved in antiviral responses to alphavirus infection (protein kinase R [PKR]; Barry et al., 2009; Gorchakov et al., 2004; Ryman et al., 2002) and IFN signaling (IRF1, IRF9, STAT2; Nair et al., 2017; Farmer et al., 2013; Bhalla et al., 2019) led to a significant increase in the number of virus-positive cells relative to a nonsilencing control shRNA (shNSC). Due to the high experimental variability observed with lentivirus transduction, we chose a subset of genes for additional validation using a secondary screen. These targets were chosen based on several criteria, including those with z-scores >2-fold, genes for which several individual shRNA sequences were detected as positive hits, genes encoding proteins predicted to be involved in RNA interactions, and targets that have not previously been explored in the context of alphavirus replication. In total, we selected 148 unique shRNAs targeting 53 genes to perform the validation screen. As a positive control, we also included an shRNA against STAT2. The validation screen was performed as described for the primary screen, and fold increase in VEEV E2-positive cells (Q2) of the transduced population (Q2+Q3) relative to shNSC was calculated (Fig. 1A, B; Supplementary Table S5). Of the 53 genes, knockdown of 24 of these resulted in a >3-fold increase in VEEV-positive cells. Knockdown of 49 of these genes resulted in a >2-fold increase in VEEV-positive cells; however, only 32 of these genes exhibited a >2-fold increase for multiple unique shRNAs (Fig. 1A). Notably, we identified several genes and pathways that have previously been implicated in restricting the replication of other alphaviruses, including SNX5 (Schuchman et al., 2018) and the ISG15 pathway [HERC6, UBE2L6 (Fig. 1B, top row) (Werneke et al., 2011; Zhang et al., 2007; Li et al., 2017)]. We also identified several IFIT proteins [IFIT1 (Hyde et al., 2014), IFIT2, and IFIT3] as restriction factors for VEEV (Fig. 1B, bottom row).

IFIT2 inhibits VEEV replication in vitro and pathogenesis in vivo. HeLa cells were transduced with lentiviruses encoding gene specific or nonsilencing control shRNAs. Cells were treated with 10 IU/mL of IFN-β for 6 h, infected with VEEV TC83 at a MOI of 1, and fixed and immunolabeled for VEEV E2 and analyzed by flow cytometry 24 h postinfection (hpi). Lentivirus (shRNA) positive cells are indicated by GFP, and E2-labeled cells were counterlabeled with Alexaflour 647 secondary antibody. Summary of shRNA validation screen (53 genes) results expressed as the fold increase in VEEV-positive cells relative to the shNSC control
We have previously identified IFIT1 as a restriction factor for VEEV and other alphaviruses (Hyde et al., 2014). As IFIT2 has previously been shown to inhibit replication of several other virus families and protect from lethal neuropathogenesis in vivo (Butchi et al., 2014; Cho et al., 2013; Davis et al., 2017; Fensterl et al., 2012; Fensterl et al., 2014; Wetzel et al., 2014) and to be upregulated in the brains of mice following neurovirulent Sindbis virus (SINV) infection (Johnston et al., 2001), we decided to further explore the role of IFIT2 in restriction of VEEV replication. To validate our in vitro findings in an in vivo model of pathogenesis, we infected WT and Ifit2 knockout (Ifit2−/− ) mice with VEEV ZPC738 and monitored animals for survival (Fig. 1C). We observed a modest but significant decrease in the average survival time of Ifit2−/− mice infected with ZPC738 relative to WT mice (P = 0.0138), indicating that in addition to inhibition of replication in vitro, IFIT2 plays some role in VEEV restriction in vivo.
The VEEV 3′UTR is a target of IFIT2
Like IFIT1, IFIT2 also possesses RNA-binding activity (Yang et al., 2012) and has been proposed to exert its antiviral activity via a mechanism dependent on this property. Studies from our lab and others have shown that IFIT1 binds and restricts RNAs containing a 5′7-methylguanisine cap (m7G; cap 0), but not 2′-O-methylated cap structures (cap 1) (Hyde et al., 2014; Pichlmair et al., 2011; Szretter et al., 2012; Kimura et al., 2013; Abbas et al., 2017). In contrast, the RNA targets of IFIT2 are poorly defined. Previous studies have demonstrated binding specificity of human IFIT2 for poly (AU) RNA as well as RNAs containing AU-rich elements (AREs) but not polyA, polyU, or GC-rich RNA (Yang et al., 2012). However, whether any AU-rich sequence is sufficient for recognition and binding by IFIT2, or whether other RNA motifs or surrounding sequences influence binding specificity is unknown. Analogous to cellular mRNAs, many +ssRNA viruses also contain AU-rich regulatory elements in the 3′ end of their genomes. As such, we speculated that the alphavirus 3′ terminus that contains AU-rich sequences is a likely target of IFIT2. To explore this hypothesis, we first performed a sliding window analysis of the VEEV TC83 genome to identify regions with AU-rich sequences (Fig. 2A). We used a Z-score cutoff of 2.32 (representing the 99th percentile) to identify regions containing significantly high AU content (average = 50.2%) and observed multiple regions in the genome with high AU content, including several genes in open reading frame 1 as well as the 3′UTR. As we predicted, the AU content of the 3′UTR, particularly the very 3′ end, was highest among all the regions identified (Z-score 2.5–4.2; Fig. 2B). This is consistent with previous studies demonstrating IFIT2-binding specificity for AU-rich sequences (Yang et al., 2012) and proposed interactions with AREs in cellular RNAs (Berchtold et al., 2008).
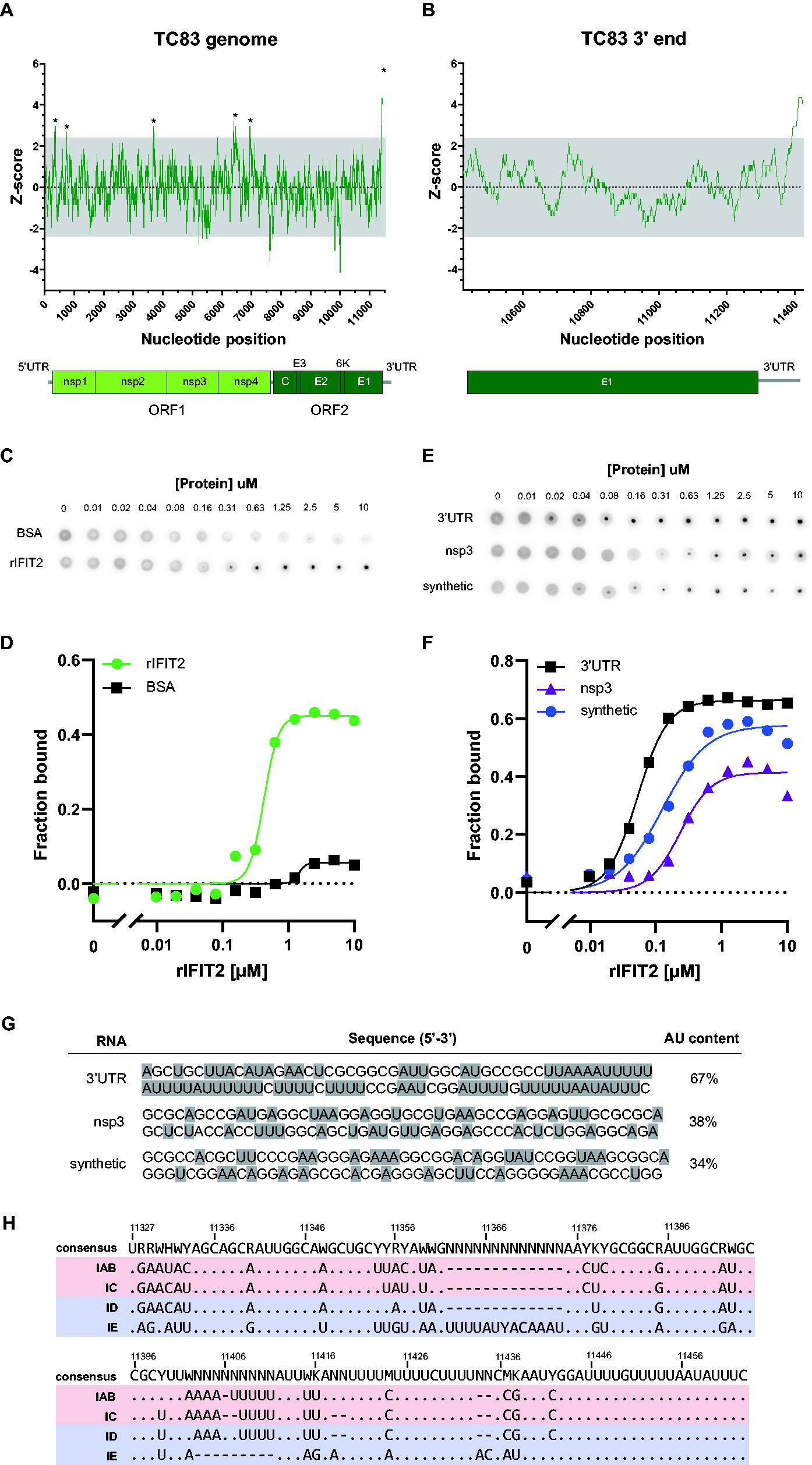
IFIT2 binds the VEEV 3′UTR. The VEEV TC83 genome nucleotide content was analyzed using sliding window analysis (window = 50 nucleotide, step = 1 nucleotide), and the Z-score of the AU content graphed
To determine whether IFIT2 indeed binds the VEEV 3′UTR, we generated recombinant mouse IFIT2 (rIFIT2) and performed Differential Radial Capillary Action of Ligand Assay (Roelofs et al., 2011) to measure interaction of IFIT2 with VEEV TC83 3′UTR RNA (Fig. 2C). Analogous to filter binding assays, DRaCALA relies on the propensity of nitrocellulose to bind proteins but not small molecules such as RNA. When protein–ligand mixtures are applied to dry nitrocellulose membranes, protein and protein-bound ligands are immobilized at the site of application, whereas unbound ligand diffuses freely through the membrane via capillary action. We in vitro transcribed and P32 end-labeled RNA corresponding to the last 200 nucleotides of the TC83 genome that constitutes the 3′UTR and 79 nucleotides of upstream sequence and incubated radiolabeled RNA with serial dilutions of rIFIT2 or BSA as a negative control. Protein–RNA mixtures were then applied to nitrocellulose and the fraction of bound RNA quantified (Fig. 2D) (Roelofs et al., 2011). We chose to examine ligand binding using an RNA consisting of additional sequence upstream of the 3′UTR as the nucleotides at the start of the 3′UTR are predicted by RNAfold (Denman, 1993) to participate in base-pairing and therefore would be disrupted by examining binding to the 3′UTR sequence alone. In these assays, we observed significantly higher binding of rIFIT2 to TC83 3′ RNA (Kd = 0.424 ± 0.051) as compared with BSA that exhibited minimal binding to RNA (Kd = 1.433 ± 0.028). Notably, the affinity of rIFIT2 for the VEEV 3′UTR is ∼10-fold lower than that which we previously observed for IFIT1 and the VEEV 5′UTR (Hyde et al., 2014), suggesting significant differences in affinity of these proteins for their respective RNAs, which likely impacts their biological activities.
To validate whether the AU content of the 3′ RNA is important for determining specificity of IFIT2 binding to RNA, we performed additional DRaCALAs comparing binding of rIFIT2 to 3′ RNA and 2 additional RNAs with lower AU content (Fig. 2E–G). The first of these corresponds to a region in nsp3 (38% AU), and second, a synthetic RNA derived from the plasmid (nonviral sequence) encoding the TC83 infectious clone (34% AU). In these experiments, we synthesized, labeled, and purified ∼100 nucleotide long RNAs as described above. Shorter RNAs were used for this experiment, as longer RNAs tended to have a higher AU content that were not significantly different from the 200 bp RNA fragment encoding the VEEV 3′ (Fig. 2C, D). In these experiments, we observed significantly higher binding of rIFIT2 to TC83 3′ RNA (Kd = 0.053 ± 0.030) as compared with either nsp3 RNA (Kd = 0.236 ± 0.069) or synthetic RNA (Kd = 0.126 ± 0.050). We observed increased binding of rIFIT2 to the 100 bp 3′ RNA as compared with the 200 bp RNA, as observed by an 8-fold decrease in the measured Kd of the shorter RNA. This may be explained by the slightly lower AU content of the 200 bp RNA (62%) as compared with the 100 bp RNA (66%), although we cannot rule out that additional structures in the 200 bp RNA may modulate IFIT2 binding. Notably, we observed decreased binding of rIFIT2 to both the nsp3 and synthetic RNAs, which exhibited a 4.5-fold and 2.4-fold increase in Kd relative to 3′ RNA, respectively (compare blue and purple lines in Fig. 2F). Interestingly, we observed that the binding affinity of IFIT2 for the nsp3 RNA was lowest, despite the fact that the AU content of this RNA is slightly higher than the synthetic RNA. At present, it is unknown what the exact determinants of IFIT2-RNA binding are (e.g., AU track length and RNA structure), and we are currently exploring this further. Overall, our binding data show that IFIT2 preferentially binds AU-rich RNA and suggests that other factors such as exact primary sequence and secondary structure may also modulate the specificity of binding.
Changes in the VEEV 3′UTR sequence alter restriction of VEEV replication by IFIT2 in vitro
To determine whether 3′UTR sequences are conserved across different VEEV species, we compared full-length and partial 3′UTR sequences from 136 strains representing 4 major VEEV subtypes (IAB, IC, ID, and IE) (Fig. 2E; Supplementary Fig. S1). As seen with TC83, we observed several stretches of polyAU sequence throughout the 3′UTR, as is also seen in many +ssRNA viruses; 3′UTR sequences from IE subtypes were considerably different from IAB, IC, and ID sequences, consistent with the fact that IE subtypes are more evolutionarily divergent from these other subtypes (Oberste et al., 1996; Oberste et al., 1999). However, notably we observed that the 3′UTR sequences of different VEEV strains contained multiple nucleotide variations. Although we did not identify a single mutation or groups of mutations that were exclusively found in viruses belonging to a single subtype (except for the more divergent IE viruses), (Fig. 2E, bottom sequence), several mutations appeared more frequently in viruses belonging to a given subtype. Therefore, we speculated that 3′UTR nucleotide variations present in individual subtypes may lead to alteration in the biological functions of viral 3′UTR RNA, including susceptibility to IFIT2 binding and inhibition of replication.
In order to test our hypothesis, we introduced select mutations from IC, ID, and IE subtypes into the 3′UTR of the TC83 infectious clone (Fig. 3A; Supplementary Table S2). VEEV subtypes exhibit genetic diversity and can be divided into different lineages, with epizootic strains being further classified into clades (Weaver et al., 2004). We chose several different 3′UTR sequences in order to capture the sequence variation observed, particularly within the ID subtype, which encompasses several major lineages and several epizootic clades (Supplementary Fig. S1; Supplementary Table S6). We compared replication kinetics of 3′UTR chimeras in primary MEF derived from WT and Ifit2−/− mice (Fig. 3B–E). MEF were mock or IFN-β pretreated for 12 h to induce IFIT2 expression and infected at a MOI of 0.01. Cell culture supernatant was harvested at 1, 6, 12, 24, 36, and 48 hpi and infectious virus quantified using FFA. When we compared replication of WT and mutant viruses in IFN-β pretreated samples, we observed that replication of TC83 and TC83/IC-3′UTR viruses (Fig. 3B, C) was consistently higher in Ifit2−/− cells as compared with WT (>1 log) though not statistically significantly (IAB WT versus KO, P = 0.3553), although differences in replication of IC in WT versus KO cells were approaching significance (0.0845). In contrast, we observed identical replication kinetics of TC83/ID-3′UTR and TC83/IE-3′UTR mutant viruses in IFN-stimulated WT and Ifit2−/− cells (Fig. 3D, E), suggesting that these viruses are resistant to the antiviral activities of IFIT2. When comparing replication of all viruses in KO cells pretreated with IFN, no biological or statistical significance was observed (Fig. 3G; compare red shaded lines). When we compared replication of all 4 viruses in WT IFN pretreated cells (Fig. 3G; compare black and gray shaded lines), we observed significant differences between epizootic and enzootic 3′UTR chimeras (IAB versus ID, P = 0.0061; IAB versus IE, P = 0.0019; IC versus IE, P = 0.0184) except for IC versus ID, although this difference was approaching statistical significance (IC versus ID, P = 0.0577). Notably, replication kinetics and viral titers in mock-treated samples were near identical for all viruses (compare solid lines in Fig. 3B–E), indicating that (1) this phenotype is IFN dependent and (2) that the observed IFIT2 phenotype is not due to replication defects or advantages caused by introduction of these mutations. Therefore, we conclude that the differential replication observed between IAB and IC versus ID and IE mutants can be attributed to the specific activities of IFIT2. Of note, TC83/IC-3′UTR and TC83/ID-3′UTR mutant viruses differ only by a single nucleotide (U-to-C mutation at nucleotide 11366; compare Fig. 3C, D), suggesting that the differences in IFIT2-mediated restriction could potentially be driven by changes in 3′UTR RNA structure and not primary sequence. This is supported by findings that the primary sequence of the IFIT2 ligand (AU-rich RNA) is somewhat broadly defined. Although higher overall AU content clearly coincides with greater IFIT2 binding (Fig. 2F, G), IFIT2 is still able to bind RNAs with variable AU content and length of AU-rich tracts. As such, we hypothesize that it would be unlikely that a single nucleotide change would significantly alter the linear IFIT2 recognition motif sufficiently to result in a significant change in IFIT2 binding or IFIT2-dependent viral replication, particularly given that the position of the nucleotide change in IC versus ID does not disrupt the AU-rich tracts that lie downstream of this nucleotide (Fig. 3A). This is further supported by our previous observations that single nucleotide mutations in the alphavirus 5′UTR are sufficient to alter RNA secondary structure in a manner that prevents binding of RNA by IFIT1 (Hyde et al., 2014).
IFIT2 differentially inhibits replication of VEEV mutants encoding distinct 3′UTR sequences.
IFIT2 modulates VEEV replication in a cell type-dependent manner
We wanted to further explore the role of IFIT2 inhibition of VEEV replication in more physiologically relevant cell types. Myeloid cells including macrophages are early targets of VEEV infection in vivo (Aronson et al., 2000; MacDonald and Johnston, 2000; Gardner et al., 2008). In contrast to nonmyeloid cells (including MEF), which fail to secrete IFN-α/β following alphavirus infection due to viral-mediated transcriptional and translational inhibition (Bhalla et al., 2016; Burke et al., 2009), macrophages are significant producers of type I IFN in vitro and in vivo (Bhalla et al., 2019). To determine the role of IFIT2 in inhibition of VEEV replication in macrophages, we generated Ifit2 CRISPR Raw264.7 macrophages using a doxycycline-inducible CRISPR/Cas9 all-in-one expression vector encoding Cas9 and individual Ifit2-specific gRNAs (g13, g25, and g38). We verified Ifit2 gene KO of bulk cells by western blotting (Supplementary Fig. S2) and then compared replication kinetics of 3′UTR mutants in WT (Raw264.7-Empty) and Ifit2−/− CRISPR KO (Raw264.7-Ifit2.g13) cells (Fig. 4A–D). As macrophages basally express IFIT2 (Supplementary Fig. S3) and secrete IFN-α/β following VEEV infection (Bhalla et al., 2019), cells were not pretreated with IFN-β prior to infection to induce ISG expression. Cells were infected at a MOI of 0.01, and cell culture supernatant was harvested at 1, 6, 12, 24, 36, and 48 hpi and infectious virus quantified by FFA.

IFIT2 differentially inhibits replication of VEEV 3′UTR mutants in a cell type-dependent manner. Growth kinetics of 3′UTR mutants in WT (empty; black) and Ifit2 KO (g13; red/blue) Raw264.7
In stark contrast to our observations of virus replication in primary MEF, where Ifit2 KO resulted in increased virus replication (Fig. 3), we observed a decrease in VEEV replication in Ifit2−/− versus WT macrophages (Fig. 4B–E), suggesting that IFIT2 functions in a cell type-dependent manner to modulate VEEV replication. However, similar to MEF, we observed that changes in the VEEV 3′UTR sequence also impact IFIT2-dependency of virus replication in macrophages. In WT cells, we observed similar viral titers over the course of the infection with TC83, TC83/IC-3′UTR, and TC83/IE-3′UTR viruses (Fig. 4A, B, D; black line). However, in contrast to TC83/IC-3′UTR, which replicated similarly in both WT and Ifit2 KO cells, TC83 and TC83/IE-3′UTR mutants exhibited significantly less replication in KO cells relative to WT (P = 0.0283 and P = 0.0382, respectively; compare black and red lines in Fig. 4A, B, D). We also observed a modest (2–5-fold) but significant decrease in replication of TC83/ID-3′UTR virus in Ifit2 KO versus WT cells (P = 0.0103). Strikingly, we observed that TC83/ID-3′UTR, which differs from IC by only a single nucleotide, replicated to significantly higher titers than TC83/IC-3′UTR in both WT (20–100-fold increase; P < 0.0001) and Ifit2 KO macrophages (20–80-fold increase; P = 0.004) (Fig. 4C), suggesting that changes in the virus 3′UTR not only affect VEEV IFIT2-dependency but also impact VEEV replication in an IFIT2-independent manner. This effect was also cell type dependent, and possibly IFN-dependent, as we did not observe any difference in replication of IC versus ID mutants in WT mock-treated MEF or in IFIT2-deficient cells following IFN pretreatment (Fig. 3).
To determine whether other single nucleotide polymorphisms (SNPs) identified in subtype ID 3′UTR sequences from different VEEV lineages (Supplementary Fig. S1; Supplementary Table S6) similarly affected virus replication in macrophages, we also constructed additional TC83/ID-3′UTR mutants (U11354C; U11355C + A11360U; U11355C + U11357C + C11364U; U11354C + C11364U + U11366U; Supplementary Table S2) and compared replication of these viruses in WT and Ifit2 KO Raw264.7 cells simultaneously (Fig. 4E–H). Remarkably, all mutants exhibited near-identical replication to each other as well as the parent ID virus, suggesting that the macrophage-specific function of the 3′UTR is conserved among different ID subtypes, despite the presence of these nucleotide variations. Collectively, these data suggest that (1) IFIT2 modulates VEEV replication in a cell type-dependent manner; (2) changes in the VEEV 3′UTR alter the IFIT2-dependency of virus replication; (3) mutations in the virus 3′UTR (IC versus ID) also affect replication of VEEV independent of IFIT2, in a cell type manner; and (4) while subtype ID viruses contain numerous SNPs, the 3′UTR-dependent replication phenotype of these viruses is conserved.
IFIT2 modulates VEEV replication via a translation-independent mechanism
Several biological functions have been ascribed to IFIT2, including translational control, cancer development, and apoptosis. Multiple studies have implicated translational control to be the major mechanism by which IFIT2 controls viral replication, which can occur through sequestration of host translation initiation factors (Terenzi et al., 2006), direct binding of viral RNA (Tran et al., 2020; Yang et al., 2012), or indirectly through interaction with IFIT1 (Fleith et al., 2018; Johnson et al., 2018). To determine whether the 3′UTR-dependent replication phenotypes observed in fibroblasts and macrophages could be explained by differences in translation, we compared translation of viral reporter RNAs [VEEV TC83/FLuc/rep; (Tesfay et al., 2008; Hyde et al., 2014)] encoding mutant 3′UTRs in WT and Ifit2 −/− primary MEF and Raw264.7 cells (Fig. 5). Translation reporters consist of the firefly luciferase gene (FLuc) fused in-frame downstream of a truncated VEEV nsP3 sequence flanked at either end by the VEEV 5′ and 3′UTRs. We chose to use reporter RNAs to assess translation as this allows us to uncouple effects of IFIT2 on viral RNA translation versus RNA replication and transcription. WT and Ifit2 −/− cells were mock- or pretreated with IFN-β for 12 h and then electroporated with in vitro transcribed m7G-capped reporter RNA. Cell lysates were harvested at 30, 60, 120, and 240 min post electroporation and translation measured by quantifying FLuc activity and normalizing to total protein. As expected, IFN pretreatment of MEF significantly inhibited translation of all reporter RNAs in both WT and Ifit2 −/− cells (compare Fig. 5A, B with Fig. 5C, D), demonstrating the contribution of other ISGs (including IFIT1) to translation inhibition of viral RNA. However, we observed no significant difference in translation of reporter RNAs encoding WT or mutant 3′UTR sequences. Interestingly, translation of all reporter RNAs appeared to trend slightly higher or lower in WT and Ifit2 −/− cells under conditions of mock- or IFN pretreatment, respectively, although this was not statistically significant.

IFIT2-dependent inhibition of 3′UTR mutants is translation independent. WT and Ifit2−/−
primary MEF were mock
As we observed contrasting replication phenotypes in fibroblasts and macrophages, we also performed translation reporter assays in WT and Ifit2 CRISPR KO Raw264.7 (Fig. 5E, F), to determine whether these differences could be attributed to differential IFIT2-dependent translation in these cell types. Assays were performed as for MEF (described above); however, unlike MEF, Raw264.7 cells were not pretreated with IFN-β as these cells basally express IFIT2 (Supplementary Fig. S3). When we compared translation of reporter RNAs in these cells, we observed a modest (∼2–4-fold) but significant decrease in translation of all reporter RNAs in WT versus KO cells, consistent with regulation role for IFIT2 in global translation regulation. However, we observed no difference in translation between reporter RNAs in either WT or KO cells, with the exception of repTC83/IE-3′UTR, suggesting that 3′UTR-dependent differences in translation do not account for our observed replication phenotypes in either fibroblasts or macrophages. Interestingly, we observed up to a 5-fold (WT) or 7.7-fold (Ifit2 −/−) increase in translation of repTC83/IE-3′UTR relative to other reporters, specifically in macrophages but not fibroblasts. Of note, we observed no difference in translation of either IC or ID in either cell type, despite the fact that the single nucleotide difference between these RNAs accounts for significant differences in replication in both cell lines (Figs. 3, 4). Collectively, our translation reporter data suggest that IFIT2 plays a modest role in global regulation of cellular translation in fibroblasts and macrophages, consistent with previous reports of the role of IFIT2 in translation inhibition and regulation (Terenzi et al., 2005; Terenzi et al., 2006). However, the mechanism by which IFIT2 inhibits (MEF) or promotes (Raw264.7) replication of 3′UTR mutants appears to be independent of viral RNA translation. Given that IFIT2 has been shown to bind to and regulate translation of cellular mRNAs (Berchtold et al., 2008; Tran et al., 2020), it is possible that IFIT2-dependent translation inhibition of a subset of cellular transcripts could explain these observations. Further studies are necessary to elucidate the molecular mechanism by which IFIT2 regulates replication of VEEV and how the viral 3′UTR contributes to replication, innate immune evasion, and pathogenesis.
Discussion
In this study, we used an shRNA screening approach to identify VEEV restriction factors and identified IFIT2 as a novel viral restriction factor of VEEV replication and pathogenesis. We identified that IFIT2 specifically binds to the 3′UTR of the VEEV genome, which contains AU-rich elements, a broadly defined motif abundant in both cellular and +ssRNA viral RNA. This is consistent with previous studies of human IFIT2, which has been shown to bind AU-rich RNA sequences (Yang et al., 2012) as well as AREs found in mouse transcripts (Berchtold et al., 2008). When we compared 3′UTR sequences from different strains of VEEV, we observed tremendous diversity in their sequences. We also noted that the 3′UTRs of viruses belonging to different subtypes (IAB, IC, ID, and IE) of VEEV were distinct and speculated that these sequence differences may confer functional changes in 3′UTR RNA that affect replication and innate immune evasion. To test this, we compared replication of IAB, IC, ID, and IE 3′UTR mutants in WT and Ifit2 −/− primary fibroblasts, as well as macrophages which are early targets of VEEV infection in vivo and important producers of type I IFN (Bhalla et al., 2019).
We made several striking observations in these cell lines. First, we observed opposing IFIT2-dependent phenotypes, with an antiviral role in fibroblasts (Fig. 3) and a proviral role in macrophages (Fig. 4). Although the majority of viral pathogenesis studies to date have predominantly shown an antiviral role for IFIT2, a more recent study by Tran et al. suggests that influenza virus usurps IFIT2 to promote translation of viral mRNAs in vitro indicating a possible proviral role (Tran et al., 2020). While our observations along with others indicate cell-type specificity, our mouse infection studies with VEEV ZPC738 show that IFIT2 predominantly plays an antiviral role during VEEV pathogenesis (Fig. 1C). As TC83 is highly attenuated in C57BL/6J mice compared with VEEV ZPC738, such direct comparisons might not be possible at this time. TC83 infection causes only modest weight loss in WT C57BL/6J mice, and only minor or modest clinical manifestations; thus, this model is not sufficiently robust to distinguish and dissect modest in vivo phenotypes. As such, we are generating 3′UTR chimeras on in other VEEV backgrounds (ZPC738), which will be used to investigate this hypothesis in following studies. We also cannot exclude the possibility that other VEEV sequences outside of the 3′UTR may contribute to IFIT2-dependent phenotypes. Nonetheless, our data show that the biological function of IFIT2 during virus replication differs depending on the cell type and likely the virus as well. In addition to cell-type dependencies, the context in which IFIT2 expression occurs (constitutive versus IFN-induced) may contribute to the seemingly disparate biological functions of IFIT2. Indeed, IFIT2 has been implicated in regulation of cellular responses (translation, cell death) as well as viral inhibition, suggesting additional roles for this protein that may be independent of IFN responses. We hypothesize that interaction of iFIT2 with different cellular protein cofactors may regulate the function of IFIT2 and ultimately determine the biological activity of this protein in different cellular contexts. These differing functions could also conceivably be regulated by post-translational modifications of IFIT2.
Second, we observed that changes in the VEEV 3′UTR had a significant effect on virus replication in an IFIT2-dependent manner. While the effect of coding sequence mutations has previously been studied in the context of epizootic VEEV emergence, the role of sequence variability in the 3′UTR has not been investigated. Despite observing opposing IFIT2 phenotypes in fibroblasts and macrophages, changes in the VEEV 3′UTR affected the dependency of VEEV replication on IFIT2 in both cell types. Interestingly, the effect of some 3′UTR mutations on IFIT2-dependent phenotypes differed between these cells. For example, in MEF, we observed an IFIT2-dependent replication phenotype with TC83/IC-3′UTR (Fig. 3C), but in macrophages, we observed no significant difference in replication of this mutant in WT versus KO cells (compare with Fig. 4C). These data may suggest that other cellular factors that are differentially expressed in these cell types also impact IFIT2. Other IFIT proteins have been demonstrated to form hetero-oligomers to modulate their functions. Recent studies have shown that IFIT2 and IFIT3 interact with IFIT1 leading to enhanced cap-binding activities of this protein (Fleith et al., 2018; Johnson et al., 2018). We propose other host factors that interact with IFIT2 alter the biological properties of IFIT2, and/or IFIT2 competes with other RNA-binding proteins for binding to the VEEV 3′UTR, and the outcome of this competition is influenced by the underlying sequence and secondary structure of the viral RNA.
We were particularly intrigued by differences in replication of IC and ID 3′UTR mutant viruses. In MEF, we observed that the ID mutant was resistant to IFIT2-mediated inhibition (compare Fig. 3C and 3D) and in macrophages observed a significant increase in replication (∼20–200-fold) relative to the IC mutant (compare Fig. 4C and 4D). These mutants differ by only a single nucleotide and are derived from equine virulent (epizootic; IC) and equine avirulent (enzootic; ID) subtypes. Epizootic (IC) VEEV subtypes arise in nature via de novo mutagenesis of enzootic subtypes (ID), which continually circulate in a sylvatic transmission cycle. With the exception of a IE epizootic outbreak in Mexico in 1996 (Gonzalez-Salazar et al., 2003; Oberste et al., 1998), IC subtypes are thought to be the predominant epizootic subtype and emerge in nature via mutation of ID enzootic viruses (Kinney et al., 1992; Powers et al., 1997; Wang et al., 1999; Wang et al., 2001; Weaver et al., 1992). Previous studies have shown that acquisition of mutations in the viral attachment protein (E2) is essential for conferring this epizootic phenotype, which is characterized by increased type I IFN resistance (Greene et al., 2005a; Greene et al., 2005b; Powers et al., 1997; Wang et al., 1999), as well as high titer viremia and pathogenesis in equines, which serve as amplification hosts during epizootic episodes (Greene et al., 2005b). While these mutations are critical for emergence of epizootic VEEV, comparative analysis of epizootic and enzootic sequences reveals that epizootic viruses also acquire other mutations elsewhere in the genome, including the 3′UTR (Supplementary Fig. S1). Significantly, a high proportion of these mutations are synonymous (Supplementary Fig. S1). While many of these mutations can likely be explained by divergent evolution due to geological constraints, we speculate that some mutations may confer biological properties (e.g., type I IFN resistance) that, although not essential, may aid in the emergence of epizootic subtypes. Thus, we speculate that acquisition of mutations that alter the function of underlying diversity in the sequences of 3′UTR may contribute to emergence of epizootic VEEV by allowing the virus to overcome the host innate immune response or by altering virus replication properties in specific intrahost niches and cell types (e.g., macrophages). Our observations that the 3′UTRs of epizootic and enzootic viruses alter the replication properties and IFIT2 dependency of VEEV lend support to this hypothesis. Furthermore, we have recently shown that RNA structures in E1 of enzootic versus epizootic VEEV alter replication fitness in macrophages (Hickson and Hyde, 2024), which are early targets of infection in vivo (Bhalla et al., 2019; Gardner et al., 2008). These studies collectively support the hypothesis that RNA sequences and secondary structures contribute to epizootic emergence in nature.
Multiple studies have demonstrated a role for IFIT2 in regulation of viral and cellular translation, either through sequestration of translation factors or through direct binding to viral RNA (Poddar et al., 2024; Terenzi et al., 2005). We observed a small decrease in translation of all reporter RNAs in Ifit2 KO versus WT cells, consistent with a role for IFIT2 in global RNA translation. However, no significant differences were observed between mutant 3′UTR reporter RNAs in either WT or KO cells. Interestingly, we observed a significant increase in translation of IE 3′UTR reporter RNA relative to the other VEEV 3′UTRs, specifically in macrophages. These data would suggest that mutations in the IE 3′UTR enhance viral RNA translation specifically in macrophages. This again bolsters our hypothesis that cell-type specific host factors expressed in macrophages are critical for determining VEEV replication; however, how this enhanced translation would impact replication and pathogenesis of IE subtypes in vivo is at present unclear. Nonetheless, these data show that our observed IFIT2-dependent replication phenotypes of 3′UTR mutants cannot be explained by differences in translation alone.
In addition to demonstrating that the VEEV 3′UTR modulates IFIT2-dependent activities, we also observed IFIT2-independent roles for the 3′UTR in VEEV replication and RNA translation. Moreover, these effects were cell type dependent, suggesting that the 3′UTR plays a crucial role in regulating replication, and possibly immune evasion, specifically in macrophages. Recent studies have demonstrated that macrophages are important early targets of VEEV infection in vivo and upon initial infection are capable of secreting significant amounts of type I IFN (Bhalla et al., 2019). This is particularly relevant as early and robust production of IFN in the periphery has been shown to limit replication and dissemination to the central nervous system of other neurotropic alphaviruses (Trobaugh et al., 2014). Thus, we speculate that changes in the VEEV 3′UTR that alter replication and innate immune responses in macrophages may have profound impacts of viral pathogenesis in vivo and is a focus of our future studies.
In summary, we identified a role for IFIT2 in restricting VEEV replication and pathogenesis in vitro and in vivo. We demonstrated that IFIT2 targets VEEV by binding to the 3′UTR and showed that changes in the VEEV 3′UTR sequence modulate the ability of IFIT2 to inhibit VEEV replication in a cell type-dependent manner. In contrast to previous studies, our data suggest that IFIT2 affects replication of VEEV 3′UTR mutants via a mechanism independent of translation. We also demonstrated an IFIT2-independent role for the VEEV 3′UTR in replication in macrophages. These findings have broad implications for the role of the alphavirus 3′UTR in the replication and pathogenesis of VEEV, as well as for the emergence of epizootic VEEV subtypes.
Footnotes
Acknowledgment
The authors thank Dr.’s Ganes Sen, Michael Diamond, and Ilya Frolov for generously providing reagents for this work.
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Funding Information
NIH grant R01 AI155416 (JLH) and UW Royalty Research Fund A172539 (JLH) supported this work.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6