
Abstract
Kawasaki disease (KD) is an acute childhood vasculitis, commonly seen in children under the age of five. Despite extensive research over the past five decades, the pathogenesis of KD remains elusive. The objective of this epigenetic reanalysis study is to delineate common pathways involved in KD using a bioinformatics approach. Array datasets from the Gene Expression Omnibus repository were extracted and subjected to analysis using the Chip Analysis Methylation Pipeline in the R statistical tool for the identification of differential methylation probes and differential methylation regions. Adaptive immune genes CD8B, RAG1, IL-7R, STAT1, and CCR7 were significantly hypermethylated in acute KD as compared to healthy controls. Gene enrichment analysis showed that genes involved in T-cell receptor activation and differentiation, antigen processing and presentation of MHC class I were hypermethylated, whereas neutrophil degranulation was hypomethylated in the acute phase of KD as compared to healthy controls. The proportion of neutrophils significantly increased, while the proportions of CD4 T-cells and CD8 T-cells decreased in the peripheral blood of children with acute KD as compared to healthy controls. Reduced proportions of CD4 T cells and CD8 T cells, as well as hypermethylation of their genes, have been observed in the peripheral blood of patients with acute KD.

Introduction
Kawasaki disease (KD) is a medium vessel vasculitis with especial predilection to coronary arteries. KD is one of the most common vasculitis syndromes that primarily affects children below five years. Approximately 15–25% of children with KD who do not receive appropriate treatment may go on to develop coronary artery abnormalities (CAAs) (Kuo, 2017; Newburger et al., 2016). With appropriate treatment, the incidence of CAAs is less than 5% in children with KD (Newburger et al., 1991).
As per the American Heart Association guidelines, diagnosis of KD is based on the presence of a set of clinical symptoms. These include fever for >5 days and the occurrence of 4 of the 5 major clinical features (diffuse mucosal inflammation, bilateral non-exudative conjunctival injection, cervical lymphadenopathy, indurative edema of the dorsum of hands and feet, periungual peeling, and polymorphic skin rashes) (Newburger et al., 2004). Patients with KD presenting with fever and <4 clinical features are diagnosed as incomplete KD. Even though the clinical signs of KD are well-known, underlying immuno-pathogenic pathways are still under exploration including the devolvement of CAAs. Further, a higher prevalence of KD in some population groups and increased susceptibility to developing disease in the siblings of KD-affected individuals, suggests a role of genetic and epigenetic factors in the etiopathogenesis of KD (Sharma et al., 2021). Therefore, there is an urgent need to understand the molecular basis of KD, which would be helpful in the early diagnosis, treatment, and disease stratification in children with KD.
Epigenetic modification such as DNA methylation plays a significant role in the regulation of gene expression without changing the DNA sequence. More recently, it has also been observed that intravenous immunoglobulin (IVIG) treatment may alter the methylation pattern and expression of genes associated with KD. For instance, IVIG administration tends to increase the methylation of genes such as matrix metalloproteinase 9 (MMP9), macrophage marker expression profiles, CD177, S100A gene family, and β-catenin in patients with KD (Huang et al., 2019; Kuo et al., 2015; Li et al., 2016). DNA methylation may serve as a link between environmental risk factors and gene transcription patterns, potentially influencing the development of KD.
We performed reanalysis of the Epigenome wide methylation dataset comprising of patients with KD (n = 12) during the acute and convalescent phases as well as healthy controls (n = 12). The present study was planned to delineate common pathways involved in KD using a Gene Expression dataset (Huang et al., 2018). Objective of this epigenetic reanalysis study is to identify differential methylation regions and genes involved in the pathogenesis of KD and use the deconvolution method to identify proportions of immune cells in the peripheral blood of children with KD.
Materials and Methods
Methylation data
We performed a search using the keywords “Kawasaki Disease” and “DNA methylation” and filtered for only Homo sapiens in the NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). Only two datasets were found matching the search query: GEO accession numbers GSE109430 and GSE84624. Raw data “.idat” files were not found in dataset GSE84624, hence excluded from further analysis. We used only the GSE109430 data for further analysis.
Bioinformatics analysis for methylation data
GSE109430 data from methylation profiling by genome tiling array was analyzed in the R software Chip Analysis Methylation Pipeline (ChAMP) package (Morris et al., 2014). Raw idat files and sample annotations were uploaded in ChAMP using champ.import function.
Low-quality probes and all probes in the SNP region were removed as part of the filtering data. Probes located in chromosomes X and Y as well as only high-quality probes (P > 0.01) were retained for analysis. Normalization of data was done for adjustment of bias in the type-II probe (Supplementary Data for before and after normalization of probes). Differential methylation probes (DMP) and differential methylation region (DMR) analysis were performed between acute phase KD (aKD) and healthy controls (HC). A similar comparison for DMP and DMR was performed separately between aKD and the convalescent phase of KD (cKD) as well as between cKD and HC. DMR analysis was performed using DMRcate and limma packages in R statistical tool (Peters et al., 2015; Smyth, 2004). The β value is the ratio of methylated intensity and the overall intensity values (sum of methylated and unmethylated probe intensities). Normalized β values of all probes for each subject were loaded in the DMRcate package to identify quantitative alteration in DNA methylation levels between the groups.
Gene set enrichment analysis (GSEA) was performed using the clusterProfiler package to map genes significantly altered in DMR for specific biological terms or pathways. Significant gene symbols from DMR were converted to Entrez gene id using the DAVID online tool (https://david.ncifcrf.gov) (Sherman et al., 2022). GSEA was performed separately for Gene Ontology (GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways using a clusterProfiler (Yu et al., 2012). A P value of <0.05 and a false discovery rate (FDR) q-value of <0.05 were considered statistically significant. Significant gene enrichment was visualized by network- and pathway-based plots using the enrichplot and pathview packages in the R Bioconductor tool, respectively.
Deconvolution method
We estimated the relative proportions of granulocytes (neutrophils), macrophages, CD4 T-cells, CD8 T-cells, B-cells, and Natural killer (NK) cells using 4 different deconvolution methods from this dataset, GSE109340. “FlowSorted.Blood.450k” is publicly available cell-sorted DNA methylation data were used as a reference library for all methods to estimate cell proportions (Reinius et al., 2012). Reference-based deconvolution methods used in this study include (i) the “estimate Cell Counts” function in the minfi Bioconductor package (Aryee et al., 2014), (ii) the robust partial correlations method in the EpiDISH (Epigenetic Dissection of Intra-Sample Heterogeneity) Bioconductor package (Zheng et al., 2019), (iii) “estimateCellProp” function in the Enmix Bioconductor package (Xu et al., 2016), and (iv) estimate Cell Counts 2 function in the Flow-Sorted.Blood.EPIC R package using the IDOL (IDentifying Optimal Libraries) algorithm (Salas et al., 2018). Proportions of each cell type should be similar in 3 of 4 deconvolution methods and considered as increased or decreased for arriving at a conclusion.
Statistical analysis
aKD and cKD were compared using a paired t-test or Wilcoxon signed rank test based on the normality of samples. KD and HC were compared using an unpaired t-test or Mann–Whitney U test based on the normality of the samples. Three groups, viz., aKD, cKD, and HC, were compared using Kruskal–Wallis test. Volcano plot, heatmap, and boxplot were generated using ggplot2 in R statistical tool.
Results
Differentially methylated sites and differentially methylated regions
A total of 485,512 probes were present in the chip array. After quality control and filtering of probes, nearly 484,100 K probes were available for analysis to examine differential methylation CpG probes between the groups. Hypermethylated and hypomethylated DMPs and DMRs between the groups are represented in Supplementary Data.
Cell proportions in KD
We estimated cell proportions using 4 different convolution methods from the normalized β value of all subjects. Among these methods, EpiDISH is consistent with any of the 3 other deconvolution methods for the estimation of the proportion of each cell type. Hence, data from the EpiDISH method are only shown. Proportions of neutrophils were increased in the aKD and decreased during the cKD as compared with aKD (Fig. 1).
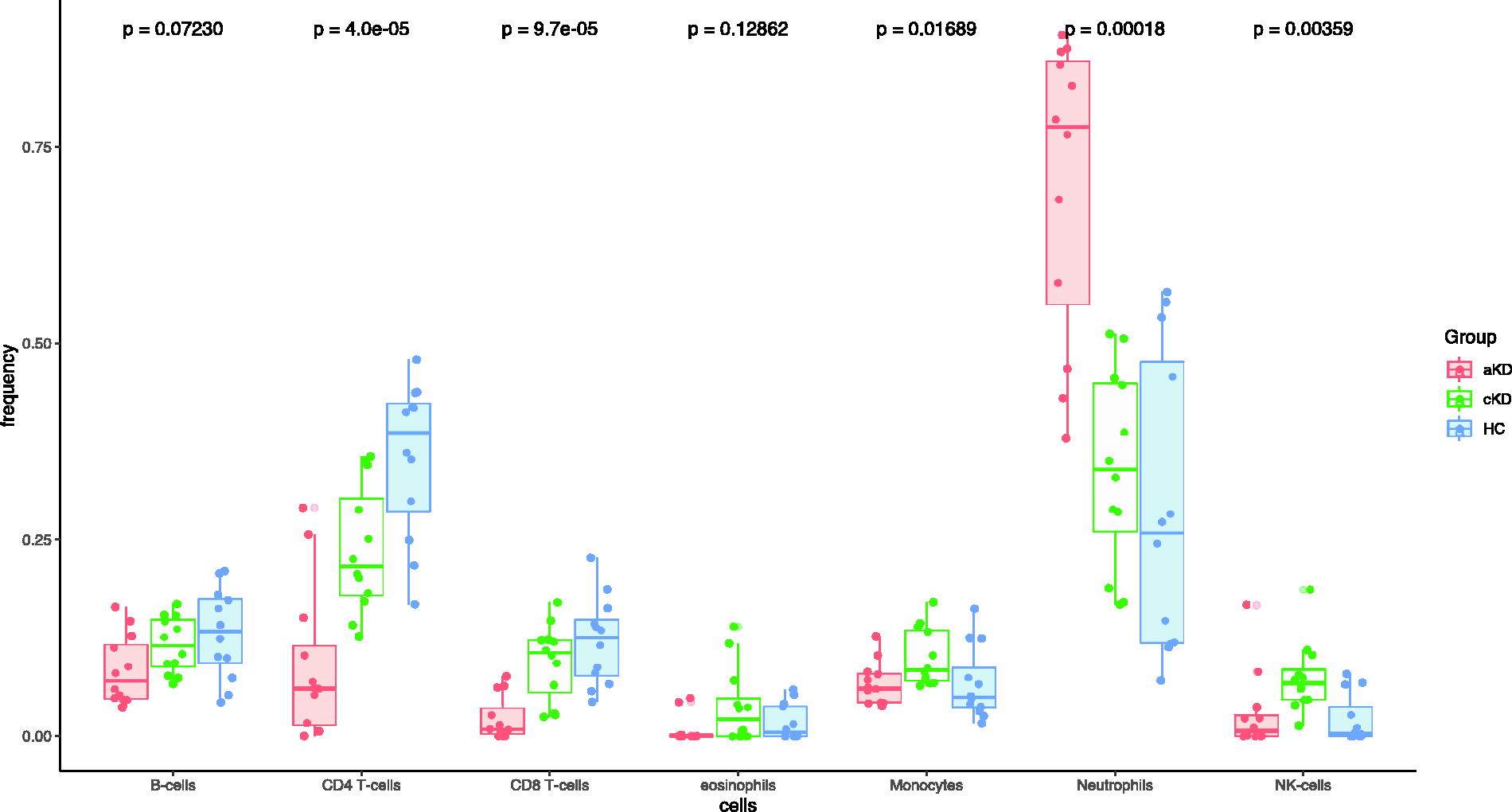
Proportions of immune cells in the blood of patients of KD during acute and convalescent phase of disease and healthy controls estimated by EpiDISH deconvolution method. Each dot represents each subject (n = 12 in each group) in the study.
Proportions of CD4 T-cells, CD8 T-cells, and B-cells were lower in aKD as compared with HC. These cells were increased to levels similar to that of HC during the cKD. Unlike CD8 T-cells, CD4 T-cells were present in low frequency in both the phases viz., aKD and cKD, in comparison to HC. The proportion of NK cells was comparable between the aKD and HC; however, a significant increase was observed in cKD.
Among the four deconvolution methods used for comparison, statistically significant difference in monocytes was only observed with EpiDISH analysis. However, no such significant difference was observed on combined analysis.
Differential methylation regions in aKD
Genes involved in neutrophil degranulation were significantly hypomethylated in patients with aKD compared to HC (Supplementary Data). However, these genes were hypomethylated but did not reach statistical significance in patients with cKD as compared with HC. There were significant hypermethylation of genes involved in adaptive immune response in aKD compared to HC and cKD.
Genes, HLA-E and LTA were significantly hypermethylated in terms of probe numbers and length in aKD (Fig. 2). Other adaptive immune genes, CD8B, RAG1, IL-7R, STAT1, CCR7, CD3G, PRF1, LCK, HLA-B, and ZAP70, were significantly hypermethylated in aKD as compared with HC (Fig. 3). These genes were hypomethylated when the patients went to the remission phase of the disease.
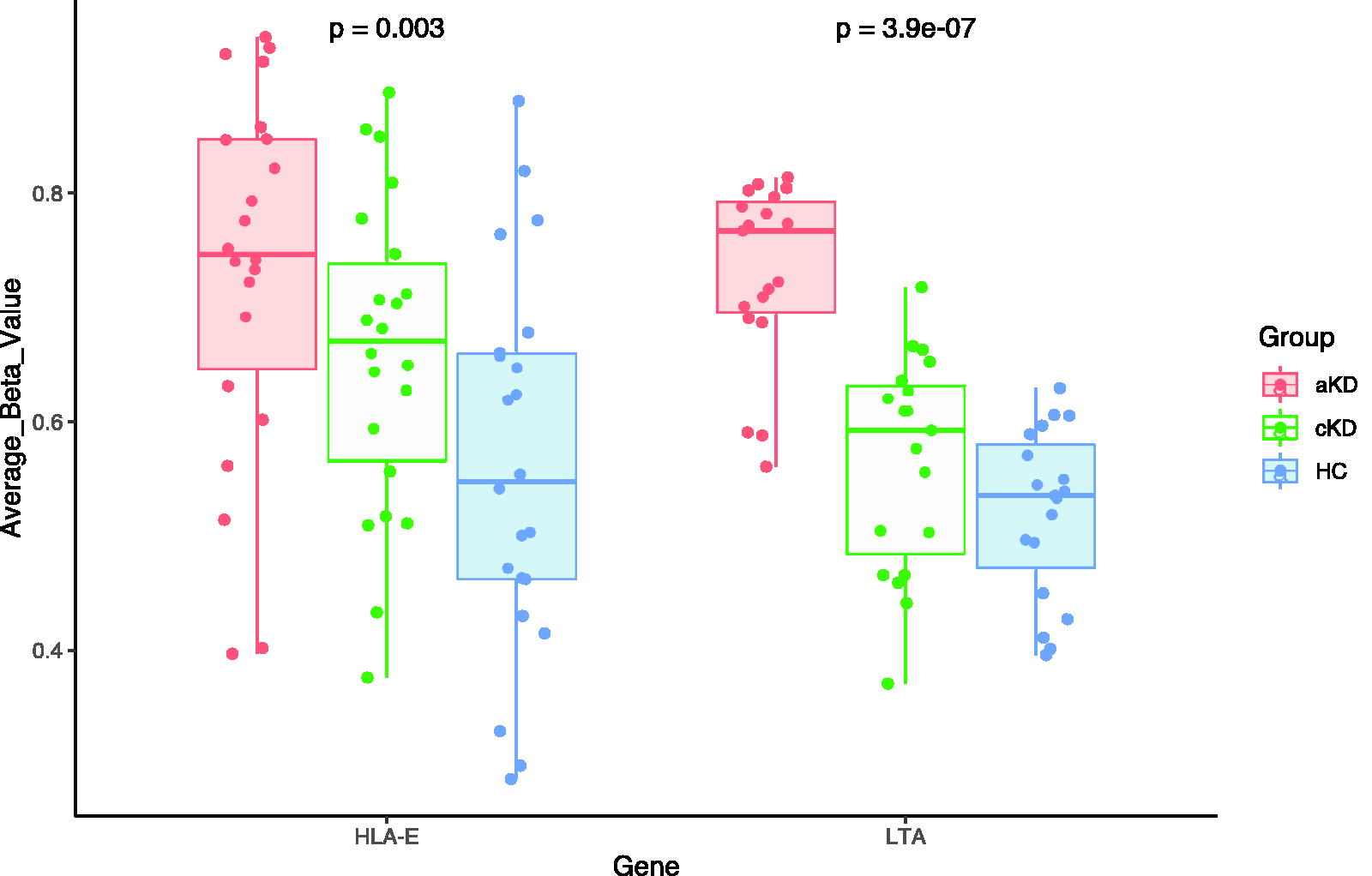
HLA-E and LTA were hypermethylated in aKD as compared with cKD and HC. Each dot represents each CpG sites measured within the gene.
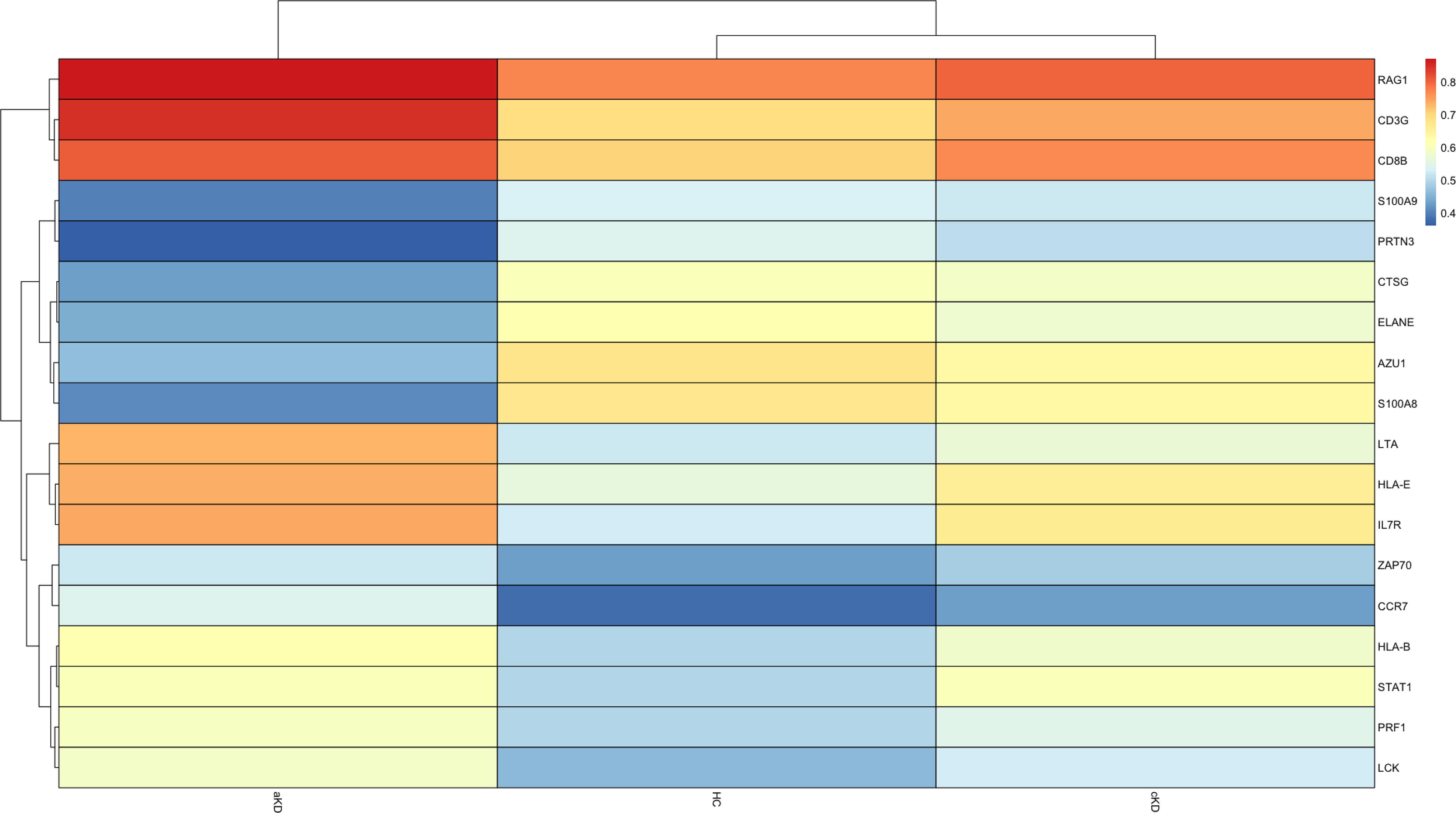
Heatmap showing hypomethylated and hypermethylated genes in aKD, cKD, and HC. Genes involved in neutrophil degranulation were hypomethylated and genes of adaptive immune response were hypermethylated in aKD as compared to cKD and HC.
DMR-associated functional pathways in KD
We identified functional pathways from average β values of 3 groups, viz., aKD vs. HC, aKD vs. cKD, and HC vs. cKD. These functional pathways were annotated in GO and KEGG databases using the genes and average β values of a particular region for the individual groups. GO analysis of the DMRs that were significant between aKD and HC revealed hypermethylation of genes involved in T-cell activation, T-cell differentiation, and T-cell selection (Fig. 4).
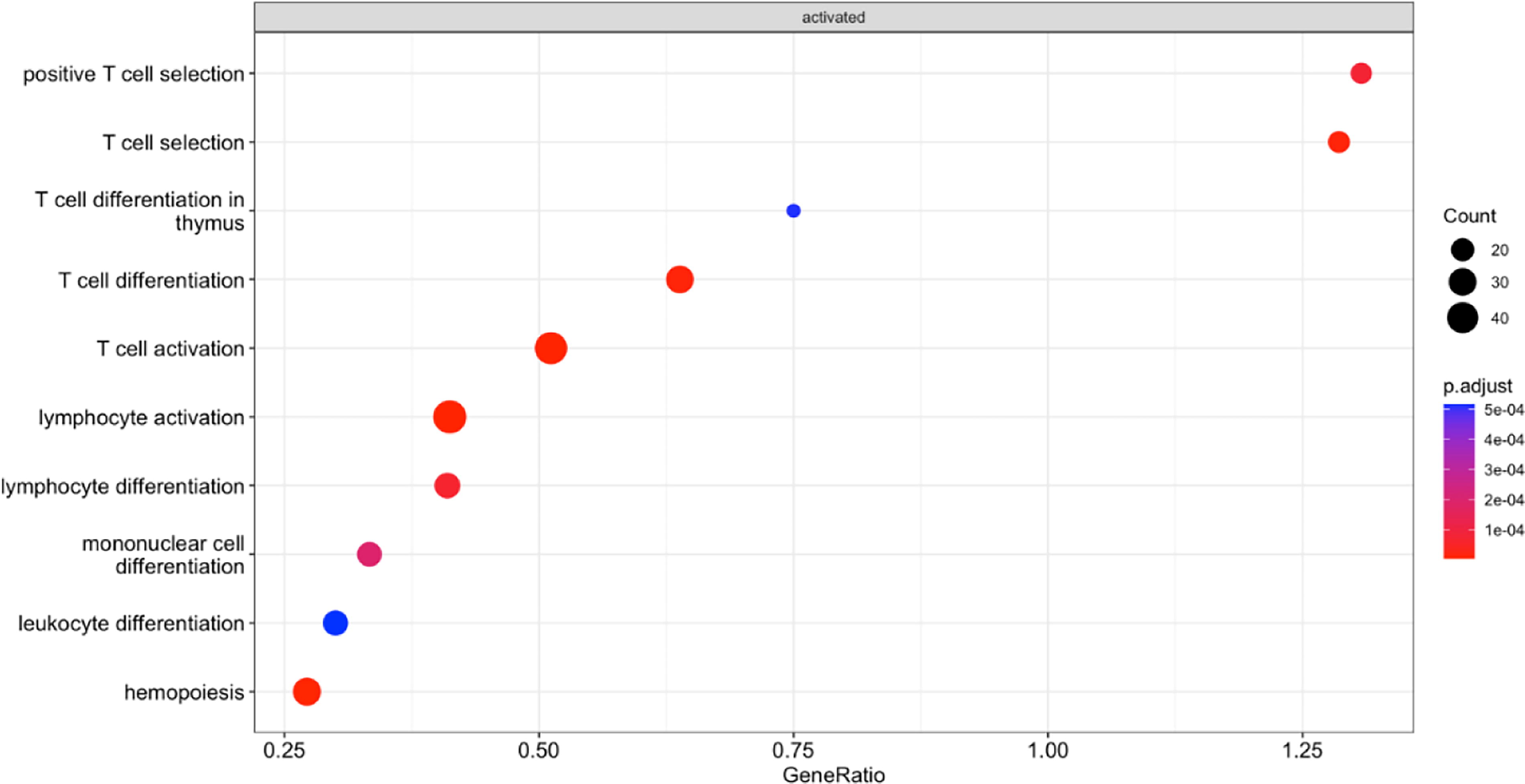
GO Gene enrichment analysis (GSE) from significant DMRs showed genes involved in adaptive immune responses were hypermethylated in aKD on comparison between aKD and HC.
Genes involved in innate immune response and immune response to bacterium were hypermethylated on comparison between aKD and cKD (Supplementary Data). No significant pathways were detected on GO analysis between cKD and HC at q-value <0.05. KEGG analysis of the DMRs that were significant between aKD and HC demonstrated an enrichment in the pathways associated with cytokine-cytokine receptor interaction, human T-cell leukemia virus 1 infection, Epstein-Barr virus infection, the JAK-STAT signaling pathway, and Th17 cell differentiation (Fig. 5 and Supplementary Data).
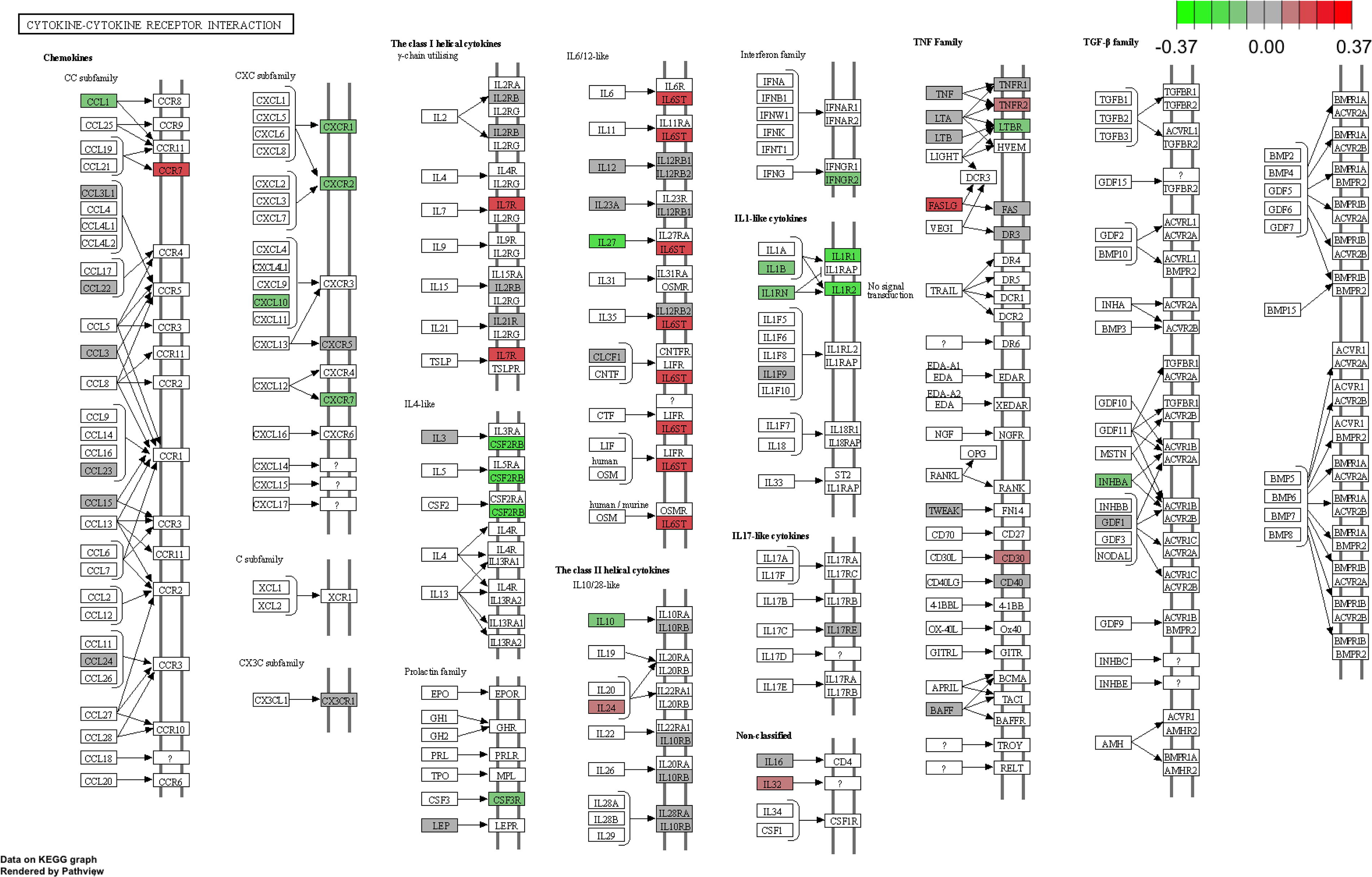
Pathway plot hypomethylated (green) and hypermethylated genes (red) involved in cytokine—cytokine receptor interaction in aKD compared to HC.
KEGG GSE analysis between aKD and cKD showed genes involved in T-cell receptor signaling were hypermethylated, and genes involving transcriptional regulation in cancer, cytokine and cytokine receptor interaction, the IL-17 pathway, and neutrophil extracellular traps were hypomethylated in aKD (Supplementary Data). Genes involved in viral myocarditis, glutamatergic synapse, purine metabolism, and Staphylococcus aureus infection were hypomethylated in cKD as compared to HC (Supplementary Data).
Discussion
KD is one of the commonest childhood vasculitis. To identify markers that are conducive to early diagnosis and small molecules with potential therapeutic effects, we performed bioinformatic analysis of GSE109430 datasets using a variety of tools. Our reanalysis of the genome-wide methylation dataset showed hypermethylation of genes involved in adaptive immune responses and hypomethylation of genes involved in neutrophil degranulation in aKD. However, there was no difference in the DNA methylation status of these genes between cKD and HC. Deconvolution analysis showed neutrophils were increased in aKD as compared with HC. NK cells and monocytes were increased in cKD as compared with HC. Proportions of CD4 T-cells and CD8 T-cells were lower in both aKD and cKD as compared with HC.
Beyond the unresolved mystery of what could trigger the disease and what leads to the development of CAAs, the most crucial clinical characteristic of acute KD, i.e., overt immunological response, has contributed to understanding its pathogenesis and etiology. Studies have demonstrated a significant role of the innate immune system in the pathogenesis of KD; however, owing to the heterogeneity and variability of lymphocytes, the role of the adaptive immune system is generally overlooked. Further, various genome-wide association studies also identified several risk loci, thereby supporting the involvement of dysregulated immune response in the pathogenesis of KD (Yan et al., 2013).
Neutrophils have been known to play a significant role in the innate immune processes in KD. We observed significant hypomethylation in several genes (AZU1, CTSG, ELANE, PRTN3, and S100A8) associated with neutrophil degranulation in aKD patients. Similar results were shown by authors of dataset GSE109430. Further, proportions of neutrophils were increased in aKD and decreased in cKD. Earlier studies of using this dataset highlight neutrophils expressing high levels of S100A proteins and leading to transendothelial migration (Huang et al., 2018). Additionally, there is an increased expression of macrophage activation markers in aKD (Guo et al., 2020). We also found that the proportion of macrophages was increased during aKD. Thus, neutrophils and macrophages play an active role in inflammatory reactions in aKD.
The genes of adaptive immunity were hypermethylated in the aKD as compared to HC. We identified significant hypermethylation in the unique gene signatures such as HLA-E, LTA, CD8B, RAG1, IL-7R, STAT1, CCR7, CD3G, PRF1, LCK, HLA-B, and ZAP70 in the patients of KD in the acute phase (aKD). These genes were hypomethylated during remission of KD (cKD). The present finding is thereby suggestive of a decreased adaptive immune response in the aKD, which normalizes during cKD. Gene enrichment pathway analysis showed that genes mainly involved in T cell activation, differentiation, and selection were hypermethylated, testifying to the decreased adaptive immune response in the aKD.
We also observed that the CD4 T cells were lower in both aKD and cKD as compared to HC, whereas NK cells were higher in cKD as compared to HC. These findings were consistent with other clinical studies wherein decreased proportion of T cells have been reported in aKD patients (Xie et al., 2022). In a recent study, Wang et al. demonstrated similar findings with a decreased percentage of CD4 T cells both in the single-cell sequencing data as well as through the flow cytometric analysis in the KD patients as compared with the HC (Wang et al., 2021). In addition, there was a significant decrease of CD8 T cells in the aKD patients as compared with the HC. Another study using a flow cytometry-based method to quantify immune cells showed decreased circulating levels of CD8 T-cells and NK cells in KD compared to febrile controls and HC (Ding et al., 2015). A similar trend was observed in the current study, wherein the proportions of CD8 T cells were lower in aKD; however, a reversion of the CD8 T cells was observed in cKD, which was consistent with other studies in the past. For instance, Brown et al. observed infiltration of the coronary artery wall primarily with CD8 T lymphocytes in the acute phase and return in circulation in cKD (Brown et al., 2001). Murine model of KD vasculitis has shown the infiltration of CD8+ T cells in coronary artery lesions and epicardial coronary arteritis. These findings suggest a role for CD8+ T cells in the development of KD vasculitis (Noval Rivas et al., 2017). This could also be suggestive of the fact that conventional antigens could elicit an adaptive immune response resulting in KD.
Popper et al. reported decreased abundance in the expression of functional genes of CD8 T-cells in aKD (Popper et al., 2007). We also observed a significant hypermethylation of genes, perforin, HLA-E, and HLA-B, which is suggestive of reduced cytotoxic function during aKD. Similar findings with decreased perforin-expressing CD8 T-cells in aKD have been reported in the past (Ehara et al., 2010; Guzman-Cottrill et al., 2005). Expression of genes involved in T-cell activation and type I interferon response was increased in coronary arteritis of KD (Rowley et al., 2015). It has also been shown that an increased proportion of HLA-DR+ T cells among the CD4+ T-cells and CD8+ T-cells is associated with treatment resistance in patients with KD (Wakiguchi et al., 2015).
It is important to note that this study is a re-analysis of previously published datasets. Some of observations, such as neutrophils and macrophages in acute KD, were previously reported. Clinical information of patients is not available, hence phenotype association with DNA methylation, has not been performed. New findings identified include decreased adaptive immune responses along with decreased frequency of CD4 and CD8 T-cells in patients with KD before IVIG as compared to healthy subjects and post-IVIG KD children.
Conclusion
The current study has shown that both the innate and adaptive immune systems are involved in the pathogenesis of KD. While neutrophil-related genes are hypomethylated during acute-phase disease, adaptive immune cells (CD4 T- cells and CD8 T-cells) are hypermethylated. Proportions of CD4 T-cells were decreased in patients with KD both during aKD and cKD as compared to healthy controls. Proportions of CD4 T-cells and CD8 T-cells were lower in both aKD and cKD as compared with HC. Further studies would be needed for better understanding the role of T cells in the etiopathogenesis of KD.
Footnotes
Data Availability
All data analyzed in this study were obtained from a previously published article and GEO accession numbers GSE109430.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.