
Abstract
The Miconia genus is the most representative of the Melastomataceae family, and some species are commonly used in Brazilian folk medicine as anti-inflammatory agents. In this work we investigated the leaves from Miconia rubiginosa (Bonpl.) DC, using high-speed countercurrent chromatography, which yielded 11 substances (eight flavonoids, gallic acid, casuarictin, and schizandriside). Identification was achieved using nuclear magnetic resonance spectroscopy and high-performance liquid chromatography–circular dichroism–diode array detection analyses.
Introduction
M
Previous phytochemical investigations of Miconia species have resulted in the isolation of triterpenes, 8,9 flavonoids, 10,11 and quinones. 12
The isolation of the compounds present in a crude extract is often performed by repeated processes based on adsorption column chromatography. In our case, we investigate mainly polar extracts containing glycosides, which can be irreversibly adsorbed on the stationary phase or even be degraded because of the catalytic activity of some solid supports. The isolated compounds are subjected to in vivo pharmacological tests, which require fast separation and milligram to gram quantities of pure compounds. High-speed countercurrent chromatography (HSCCC) uses no solid support and has a greater maximum capacity, excellent sample recovery, efficient separation of closely related natural products, and a wider choice of solvent systems compared with conventional high-performance liquid chromatography (HPLC).
A prerequisite for the full understanding of molecular events involved in the biological activity of chiral molecules is the knowledge of their absolute configuration. 13 The pharmacological and pharmacodynamic properties of biologically active chiral compounds are often strictly related to their stereochemistry. Enantiomers usually differ in biological activities because of their disparate interactions with enzymes and other naturally occurring chiral molecules. 14 HPLC with circular dichroism (CD) detection is an excellent method for the separation and detection of these chiral compounds.
There are no reports concerning the chirality of molecules isolated from Miconia species; therefore, the scope of our analysis was to search for an efficient method to isolate in preparative scale the secondary metabolites contained in the infusion of Miconia rubiginosa (Bonpl.) DC and to establish the absolute configurations of the chiral isolates.
Materials and Methods
Reagents
All solvents used for HSCCC were of analytical reagent grade from Merck (Darmstadt, Germany). The solvents used for HPLC were of analytical grade from J.T. Baker (Phillipsburg, NJ, USA). Water was nanopure quality, obtained from a Milli-Q® purification system (Millipore Corp., Bedford, MA, USA).
Preparation of crude sample and sample solution
Aerial parts of M. rubiginosa were collected in March 2005 at Palmeiras da Serra, Pratânia, SP, Brazil, and authenticated by Dr. Luiz Fernando Rolim de Almeida from the Botany Institute, State University of São Paulo, Botucatu, SP, Brazil. A voucher specimen (number 25376) was deposited in the Irina Delanova Gemtchujnicov Herbarium of the Biosciences Institute, State University of São Paulo.
The dried leaves of M. rubiginosa (100 g) were separated, powdered, and extracted at 70°C for 10 minutes in 1 L of water, yielding an infusion.
Because of the presence of tannins, the infusion (1 L) was partitioned with ethyl acetate (3×500 mL), with the polymeric tannins remaining in the aqueous phase and the secondary metabolites transferred to the ethyl acetate phase. The ethyl acetate fraction was evaporated at 35°C under reduced pressure, yielding crude extract (1.5 g, 1.5% yield). The ethyl acetate fraction (1.5 g) was dissolved in a mixture consisting of 20 mL lower phase + 20 mL upper phase of the solvent system ethyl acetate/n-propanol/H2O (14:0.8:8 by volume).
HSCCC
The preparative HSCCC instrument used in this study was from P.C. Inc. (Potomac, MD, USA). It was equipped with a multiplayer with two coils of 1.68-mm (i.d.) polytetrafluoroethylene tubing of approximately 80 and 240 mL, respectively, with a total capacity of 320 mL. The β value varied from 0.5 at the internal to 0.85 at the external terminal, and the revolution radius was 10 cm (β=r/R, where r is the distance from the coil to the holder, and R is the revolution radius or the distance between the holder axis and the central shaft). The flow rate was controlled with a Waters (Milford, MA, USA) model 4000 constant-flow pump. The sample was injected with a injection module (P.C. Inc.) with a 20-mL sample injection loop. The coiled column was filled with the stationary phase (lower phase), then the apparatus was rotated forward at 850 rpm, and the mobile phase (upper phase) was pumped into the column in a head-to-tail direction at a flow rate of 1.0 mL/minute. After the mobile phase front emerged and the hydrodynamic equilibrium was established in the column, about 20 mL of the sample solution containing 0.75 g of the ethyl acetate fraction was injected through the injection module at a flow rate of 1.0 mL/minute. Sixty fractions of 5 mL each were collected with a Redifrac automated fraction collector (Pharmacia, Uppsala, Sweden), in approximately 5 hours. After thin-layer chromatography (TLC) analyses, fractions with similar retardation factor (R F) were combined. The procedure was repeated with the remaining 0.75-g ethyl acetate fraction.
Preparation of the two-phase solvent system
The solvent system, composed of ethyl acetate/n-propanol/H2O (14:0.8:8 by volume), was thoroughly equilibrated overnight in a separator funnel at room temperature, and the two phases were separated shortly before use.
TLC and HPLC analyses
Aliquots of the ethyl acetate and the collected fractions were analyzed on glass-backed silica gel TLC (20×20 cm, Aldrich, Milwaukee, WI, USA), with the mobile phase being CHCl3/methanol/n-propanol/H2O (5:6:1:4 by volume). The TLC plates were evaluated under ultraviolet (UV) light (254 nm) and derivatized with anisaldehyde/H2SO4 solution. 15 The infusion and the isolated metabolites were additionally analyzed using a Jasco® (Tokyo, Japan) HPLC system equipped with a PU-2089 Plus pump, a MD-2010 Plus photodiode array detector, a CD-2095 Plus CD detector, an AS-2055 Plus autosampler, and a C18 column (250×4.60 mm i.d.; particle size, 5 μm; Luna, Phenomenex, Torrance, CA, USA), and a Phenomenex security guard (4.0×2.0 mm i.d.). EZChrom Elite™ version 3.1.7 software (Agilent Technologies, Palo Alto, CA, USA) was used for control of the analytical system, data collection, and processing. The mobile phase was acetonitrile + trifluoroacetic acid (0.05%) (eluent A) and H2O + 0.05% trifluoroacetic acid (eluent B), with a linear gradient elution of A:B 13:87 (vol/vol) to A:B 23:77 (vol/vol) in 40 minutes and then to 100% A in 45 minutes at a flow rate of 1.0 mL/minute, and the effluent was monitored at 254 nm. The infusion was applied to a solid-phase extraction cartridge (20 mg, Sep Pak C18, Waters), which was preconditioned with 10 mL of HPLC-grade methanol and 10 mL of water (18 mΩ·cm). A suitable amount of sample (to load 20 mg of infusion/g of solid phase) was loaded onto the C18 cartridge, eluted with 5 mL of H2O/methanol (8:2, fraction 1), 5 mL of H2O/methanol (1:1, fraction 2), and 5 mL of methanol (100%, fraction 3), and dried using a stream of N2. The residues of the three fractions were redissolved in 2 mL of H2O/methanol (8:2, vol/vol) and filtered through a membrane polytetrafluoroethylene filter (pore size, 0.45 μm; Millex, Millipore), and 20 μL of this filtrate was subjected to HPLC-UV-diode array detection (DAD) analysis.
The chiral study was performed with a polysaccharide-derived chiral stationary phase column (Chiralcel OD-RH®; particle size, 5 μm; 250×4.6 mm) protected by a Chiralcel OD-RH guard column (particle size, 5 μm; 10×4.0 mm) (Daicel Chemical Industries, Tokyo). The CD spectra were scanned between 220 and 420 nm for the resolved enantiomers using the online CD detector. The spectra were average computed over three instrumental scans, and the intensities are presented in terms of ellipticity values.
Structural identification of the compounds
Nuclear magnetic resonance (NMR) spectra (one- and two-dimensional) were obtained in dimethyl sulfoxide (DMSO)-d6 using a Varian (Palo Alto) INOVA 500 spectrometer operating at 500 MHz for 1H and 125 MHz for 13C.
Spectroscopic data
Compound
Compound
Compound
Results
A series of experiments was performed to determine a suitable two-phase solvent system for HSCCC. Small amounts of the ethyl acetate fraction were dissolved in test tubes containing each of the following solvent systems: ethyl acetate/n-propanol/H2O (14:0.8:8 by volume), ethyl acetate/n-butanol/H2O (14:0.4:8 by volume), and hexane/ethyl acetate/methanol/H2O (1:1:1:1 by volume). The test tubes were shaken, and the system was allowed to equilibrate between the two phases. An aliquot of each phase was spotted on a TLC plate (CHCl3/methanol/n-propanol/H2O [5:6:1:4 by volume]), visualized under UV light (254 nm), and developed with anisaldehyde/H2SO4 solution. The two-phase solvent system was selected according to the partition coefficients (K) of the target components. The K values were determined by TLC analysis according to Conway. 16 The best result was obtained with the mixture of ethyl acetate/n-propanol/H2O (14:0.8:8 by volume), with the substances almost equally distributed between the two phases. The relatively high proportion of ethyl acetate in the solvent mixture and the R F values of the compounds indicate the medium polarity of the metabolites of M. rubiginosa. Thus the upper phase was chosen as the mobile phase, and the lower phase was used as the stationary phase for the HSCCC separation of the ethyl acetate fraction of the infusion of M. rubiginosa. This choice has the additional advantage that the upper phase consists largely of the volatile ethyl acetate, readily eliminated by evaporation.
Under the conditions described, the retention of the stationary phase in the HSCCC was 87%. This method led to the isolation of the compounds in approximately 3.5 hours, and the whole process was completed in 5 hours. Spectroscopic analyses allowed identification of 11 main compounds, determined by comparison with literature data: quercetin-3-O-α-rhamnopyranoside (

Secondary metabolites of M. rubiginosa.
The identity and purity of the isolated substances were verified by HPLC-CD-DAD analyses using authentic samples from a collection of our laboratory. All substances were obtained with purity >95%.
Figure 2 shows the HPLC chromatographic analysis of fraction 2 of the infusion of M. rubiginosa aerial parts. The peaks were identified by co-injection with purified metabolites.
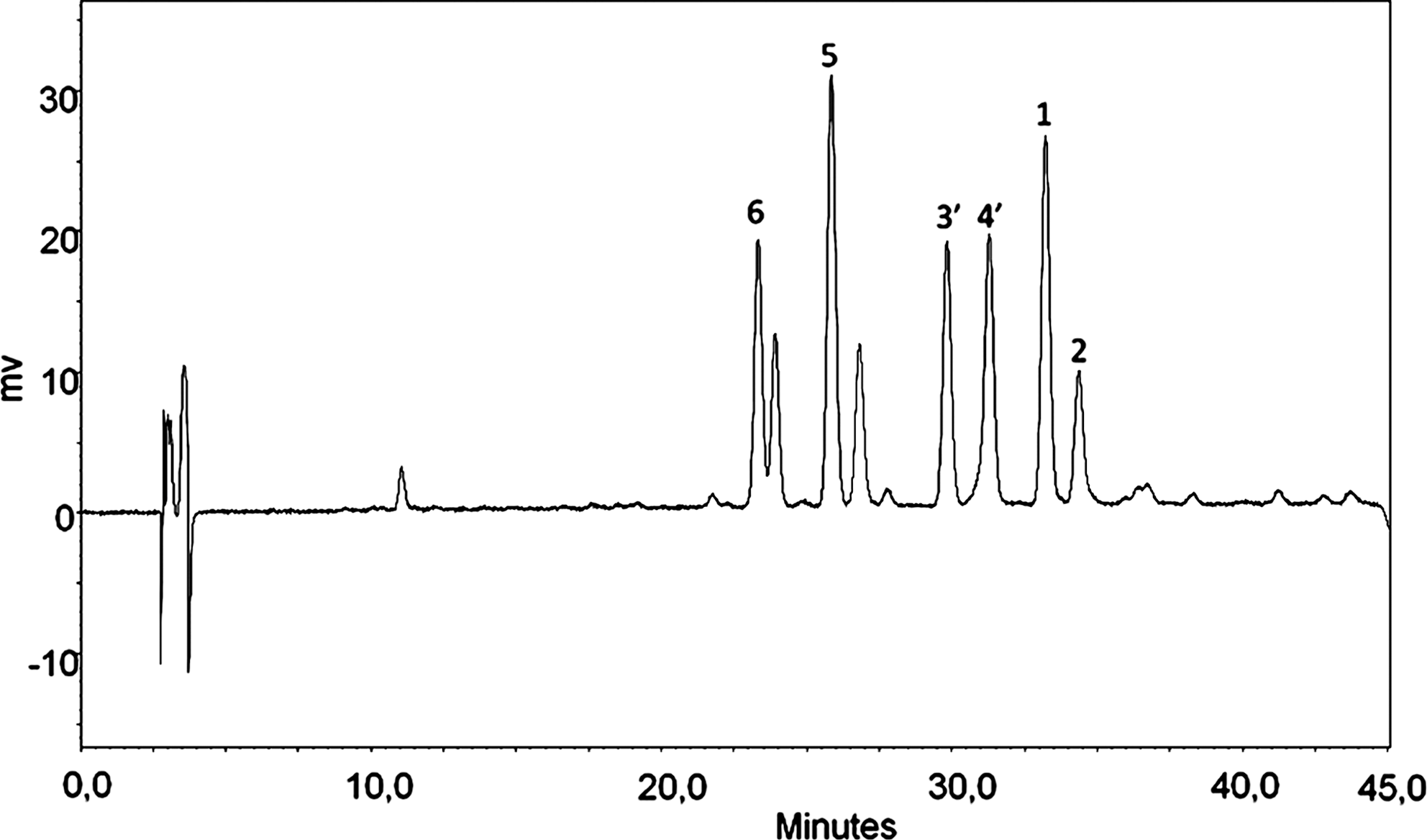
Chromatogram of the infusion of M. rubiginosa by high-performance liquid chromatography analysis: quercetin-3-O-α-rhamnopyranoside (
The compounds isolated—(–)-epicatechin (

Circular dichroism (CD) spectra of (
Discussion
Several classes of natural products have been isolated by HSCCC, including flavonoids.
23,24
Our group reported the isolation of naphthopyranone glycosides from Paepalanthus microphyllus (Family Eriocaulaceae),
25
quercetin glycosides and the biflavonoid amentoflavone from Byrsonima crassa (Family Malpighiaceae),
26
the flavones 6-methoxyluteolin-7-O-β-
The separation mechanism of HSCCC is based mainly on the differential solubility of the compound between the two immiscible phases, one flowing along the other, allowing the fast and efficient isolation of several compounds without decomposition. HSCCC, being a support-free liquid–liquid partition chromatographic technique, eliminates irreversible adsorption of the sample onto the solid support, and therefore this method has been widely used for the preparative separation of natural products. 29,30
Despite the similar polarities of the compounds, they were well separated, with the separation of the quercetin and kaempferol glycosides dependent on the total number of the hydroxyl groups. Our results showed that the conditions used provided an efficient method for the separation of the metabolites from M. rubiginosa.
Catechins are an important class of secondary metabolites. They show strong antioxidant activity and have various health-related activities, such as anticarcinogenic, anti-allergic, anti-atherogenic, antibacterial, and antiviral activities. 31 These biological activities are significantly influenced by stereochemical differences, thus justifying the determination of the chirality of these metabolites.
Catechin derivatives have two stereocenters, resulting in four diastereomers, These substances have two aromatic chromophores, namely, the benzene A- and B-rings, with the absorption bands of these chromophores found at approximately 280 and 240 nm.
32
The positive Cotton effect in the 240 nm region (L
a transition, chromophore B) and the negative Cotton effect in the 280 nm (L
b transition, chromophore A) regions indicate (2R,3R) absolute configuration, elucidating
Casuarictin (
Casuarictin (
The CD spectra of
Schizandriside (
The absolute structure of
The results of our studies clearly demonstrate the potential of HSCCC for the preparative isolation of secondary metabolites from the leaves of M. rubiginosa. In particular, preparative HSCCC, with its speedy separation and minimum solvent consumption, offers an efficient method for the separation and purification of natural products.
The compounds
Footnotes
Acknowledgments
We thank the Fundação de Amparo à Pesquisa do Estado de São Paulo for funding from the Biota-Fapesp Program and the Conselho Nacional de Desenvolvimento Científico Tecnológico for a grant to W.V. We also thank CAPES for a fellowship to J.R. and M.A.S.
Author Disclosure Statement
No competing financial interests exist.