
Abstract
Brasenia schreberi Gmel. (Cabombaceae) is an aquatic plant that grows in eastern Asia, Australia, Africa, and North and Central America. B. schreberi leaf extracts were obtained by sequential solvent extraction with dichloromethane, methanol, and water. The antioxidant potential of each extract was assessed by using the oxygen radical absorbance capacity (ORAC) assay. With this method, methanol and water extracts were found to be active with mean±standard deviation values of 7±2 and 5.1±0.5 μmol Trolox® equivalents (TE)/mg, respectively. Two major phenolic compounds, quercetin-7-O-β-D-glucopyranoside and gallic acid, were respectively isolated from the methanolic and water extracts. Both compounds exhibited antioxidant activities, in particular quercetin-7-O-β-D-glucopyranoside (ORAC value, 18±4 μmol TE/μmol). In contrast to its well-known antioxidant homologue quercetin, quercetin-7-O-β-D-glucopyranoside does not inhibit growth of human fibroblasts (WS-1) or murine macrophages (RAW 264.7). Some flavonoids have been reported to possess beneficial effects in cardiovascular and chronic inflammatory diseases associated with overproduction of nitric oxide. Quercetin-7-O-β-D-glucopyranoside possesses anti-inflammatory activity, inhibiting expression of inducible nitric oxide synthase and release of nitric oxide by lipopolysaccharide-stimulated RAW 264.7 macrophages in a dose-dependent manner. Quercetin-7-O-β-D-glucopyranoside also inhibited overexpression of cyclooxygenase-2 and granulocyte macrophage–colony-stimulating factor.
Introduction
B
The composition and structure of polysaccharides from the mucilaginous coating of the plant have been partly characterized, but few studies have been conducted on the leaves. 3,8,9 Previous studies reported that leaves of B. schreberi contain polysaccharides and unusual polyamines, such as homospermidine, norspermidine, thermospermine, norspermine, aminopropylhomospermidine, bis(aminopropyl)ethanediamine, and methylspermidine. 10,11 In the current study, we investigated the antioxidant and anti-inflammatory activities of quercetin-7-O-β-D-glucopyranoside (quercimeritrin), a major compound isolated from the leaves of B. schreberi.
Materials and Methods
General experimental procedure
Infrared spectra were recorded with a Perkin-Elmer Spectrum One FTIR using KBr pellets. Nuclear magnetic resonance spectra were recorded in deuterated dimethyl sulfoxide (DMSO) or deuterium oxide on a Bruker Avance 400 spectrometer (5 mm QNP with Z-gradient probe) operating at 400.13 MHz (1H) or 100.61 MHz (13C). The spectra were referenced to tetramethylsilane as internal standard (ppm). All high-performance liquid chromatography (HPLC) analytical investigations were carried out with an Agilent 1100 series (Agilent Technologies Canada Inc.) HPLC–mass spectometry (MS) system equipped with a vacuum degasser, a quaternary pump, an autosampler, a thermostated column compartment, and a diode array detector. Ultraviolet spectra were extracted directly from the diode array detection signal. Atmospheric pressure chemical ionization (APCI) analyses were performed with an Agilent 1100 LC/MSD quadrupole mass spectrometer; nitrogen was used as a nebulizing gas at a pressure of 35 psig, a flow rate of 12 L/min, a temperature of 350°C, a capillary voltage of 2 kV, a fragmentor voltage of 100 V, and a corona current of 15 μA. The preparative HPLC was carried out with an Agilent 1100 system equipped with an autosampler, 2 preparative pumps, a diode array detector, and a fraction collector. The accurate mass determination was carried out with an MDS Sciex QSTAR XL QqTOF MS system (Applied Biosystems) fitted with an ESI Micromass gas chromatography–time-of-flight MS system source (Waters).
Thin-layer chromatography, HPLC, and HPLC-MS conditions
The thin-layer chromatography (TLC) plates (silica gel ultrapure 250 μm, F254) were supplied by Silicycle. The TLC mobile phases were CHCl3-MeOH-H2O (26:14:3) for compounds 1 and 2, CH2Cl2-MeOH-H2O (50:25:5) for sugars, and CHCl3-MeOH (25:1) for aglycones. Phenolic compounds were detected by spraying natural products with polyethylene glycol (NP/PEG) reagent and observing them under ultraviolet light (365 and 254 nm). Other compounds were detected by using a 0.2% naphthoresorcinol in ethanol/85% H3PO4 (22:1) spray reagent and observing them under visible light. 12 HPLC analyses were carried out with a ZORBAX Eclipse XDB-C8 analytical column (5 μm, 4.6×150 mm), a flow rate of 1.0 mL/min, and a column temperature of 22°C. An injection volume of 10 μL of the sample dissolved in DMSO (2 mg/mL) was used. Preparative HPLC was performed with a ZORBAX Eclipse XDB-C8 semi-preparative column (5 μm, 9.4×250 mm) at a flow rate of 4 mL/min. Sample concentration was 50 mg/mL in DMSO. MS spectra of purified compounds were obtained through direct injection, with MeOH used as the mobile phase.
Plant material
B. schreberi was collected in August 2003 in the Saguenay region of Québec, Canada. Plants were identified by Patrick Nadeau of the Laboratoire d'Écologie Végétale of the Université du Québec à Chicoutimi, Canada. A voucher specimen (QFA-368450) was deposited in the Louis-Marie Herbarium of Université Laval, Québec, Canada. Air-dried leaves were stored at −20°C in hermetic bags for 7 months before extraction.
Extraction and isolation of compounds
The air-dried leaves of B. schreberi (100 g) were extracted for 24 hours in a Soxhlet apparatus using 250 mL of CH2Cl2. The thimble was dried in vacuum (24 hours), re-extracted with 250 mL MeOH (24 hours), dried again in vacuum (24 hours), and re-extracted with 250 mL H2O (24 hours). The organic extracts were evaporated to yield 2.8 g (3%) from CH2Cl2 and 15.3 g from MeOH (15%). The water extract was freeze-dried to yield 4.5 g (5%). The MeOH extract (14 g) was suspended in H2O (300 mL) and partitioned 3 times with 150 mL of water saturated with n-BuOH. The organic phase was decanted and evaporated in vacuum. The residue (5.9 g) was subjected to column chromatography using silica gel (200 g, 2.5×90 cm) and eluted with CHCl3-MeOH-H2O (26:14:3) to yield 8 fractions. Fraction 4 (225 mg), containing a major compound, was resubjected to silica gel column chromatography using mixtures of CHCl3/MeOH of increasing polarities as eluents. A mixture of 156 mg of compounds with Rfs of 0.5–0.8 was obtained. The major product (orange on TLC after NP-PEG treatment) was purified by preparative HPLC using the mobile phase H2O-MeOH (70:30) to give pure quercetin-7-O-β-D-glucopyranoside 1 (14 mg, 0.014% from air-dried leaves). The water extract (4.3 g) was dissolved in 300 mL H2O and was extracted 8 times with 100 mL ethyl acetate. The fractions were combined, dried over Na2SO4, and evaporated to yield 1.2 g (29%) of a brown residue, which was subjected to silica gel column chromatography (100 g, 4.5×15 cm) and eluted with the solvent gradient CHCl3-MeOH-H2O (40:10:1–10:10:1) to give 7 fractions. Fraction 2 (461 mg) was used directly for preparative HPLC separation to purify the major compound (violet on TLC after treatment) from the mixture (Rf, 0.5–0.9). Elution with H2O for 10 minutes gave pure gallic acid 2 (5 mg, 0.005% from air-dried leaves).
Quercetin-7-O-β-D-glucopyranoside (1): yellow powder. APCI-MS m/z: 463 [M–H]– and 301 [M–H–glucose]–; high-resolution electron spray ionization–MS [HR ESI-MS] (positive) m/z: 487.0844 (calculated for C21H20O12Na, 487.0852); UV
Gallic acid (2): white powder. APCI-MS m/z: 169 [M–H]– and 125 [M–COOH]–; HR ESI-MS (positive) m/z: 214.9935 (calculated for C7H6O5 − H + 2Na, 214.9932); UV
Oxygen radical absorbance capacity assay
The procedure was performed as described by Ou et al., 13 with some modifications. Oxygen radical absorbance capacity (ORAC) assay was carried out on a Fluoroskan Ascent FL™ plate reader (Labsystems), and Trolox® was used as a control standard. The experiment was conducted at 37.5°C and a pH of 7.4 with a blank sample in parallel. The fluorometer was programmed to record the fluorescence of fluorescein every 60 seconds after the addition of 2,2′-azobis(2-amidino-propane) dihydrochloride. The final results were calculated by using the differences between the areas under the fluorescein decay from the blank and the sample curves. ORAC values were expressed as μmol Trolox equivalents [TE]/mg).
Cell culture
The human fibroblast WS-1 (#CRL-1502), the murine fibrosarcoma L-929 (#CCL-1), and the murine macrophage RAW 264.7 (#TIB-71) cell lines were all obtained from the American Type Culture Collection. The WS-1 and L-929 cell lines were grown in minimum essential medium containing Earle's salts (Mediatech Cellgro®), and the RAW 264.7 cell line was grown in Dulbecco's modified Eagle's medium (Cellgro). Both media were supplemented with 10% fetal calf serum (Hyclone), 1 X solution of vitamins (Cellgro), 1 X sodium pyruvate (Cellgro), 1 X nonessential amino acids (Cellgro®), 100 IU of penicillin, and 100 μg/mL of streptomycin (Cellgro). Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Antioxidant cell assay using 2′,7′-dichlorofluorescein-diacetate
Antioxidant activity was evaluated by using 2′,7′-dichlorofluorescein-diacetate (DCFH-DA) assay as described by Girard-Lalancette et al., with some modifications. 14 L-929 cells were plated in 96-well microplates (BD Falcon) at 5000 cells per well and incubated for 48 hours at 37°C and 5% CO2. The cells were washed with 150 μL Hank's buffered salt solution (HBSS; pH, 7.4), incubated with 100 μL HBSS (pH, 7.4) containing 5 μM DCFH-DA (Sigma-Aldrich) for 30 minutes and washed again with 150 μL HBSS. To assess antioxidant activity, the cells were incubated with growing concentrations of B. schreberi methanol extract, Trolox, or quercetine, in the absence or presence of 200 μM tert-butyl hydroperoxide. The fluorescence was measured after 1 hour and 4 hours on the automated plate reader Fluoroskan Ascent FL, using an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Cytotoxicity assay
Exponentially growing cells were plated at a density of 5×103 cells per well in 96-well microplates (BD Falcon) in 100 μL of culture medium and were allowed to adhere for 16 hours before treatment. Then, the cells were incubated for 48 hours in the presence or absence of 100 μL of increasing concentrations of quercetin (Sigma-Aldrich) or quercetin-7-O-β-D-glucopyranoside in DMSO. The final concentration of DMSO in the culture medium was maintained at 0.5% (v/v) to avoid toxicity. Cytotoxicity was assessed by using the resazurin reduction test as described by O'Brien et al. 15 Fluorescence was measured on an automated 96-well Fluoroskan Ascent Fl plate reader using an excitation wavelength of 530 nm and an emission wavelength of 590 nm. Cytotoxicity was expressed as the concentration of drug that inhibited cell growth by 50%.
Nitrite oxide measurements
Exponentially growing cells were plated in 24-well microplates (BD Falcon) at a density of 2×105 cells per well in 400 μL of culture medium and were allowed to adhere overnight. Cells were then treated or not with NG-nitro-L-arginine methyl ester (L-NAME; positive control), quercetin, or quercetin-7-O-β-D-glucopyranoside dissolved in the appropriate solvents at 37°C for 24 hours. The final concentration of solvent in the culture medium was maintained at 0.5% (v/v) to avoid solvent toxicity. Cells were then stimulated with lipopolysaccharide (100 μg/mL) for 24 hours. Cell-free supernatants were collected and stored at −80°C until nitric oxide (NO) determination using the Griess reaction with minor modifications 16 : 100-μL aliquots of cell supernatants were incubated with 50 μL of 1% sulfanilamide and 50 μL of 0.1% N-1-naphthylethylenediamine dihydrochloride in 2.5% H3PO4 at room temperature for 20 minutes. Absorbance at 540 nm was then measured using an automated 96-well Varioskan Ascent plate reader (Thermo Electron), and the presence of nitrite was quantified by comparison with an NaNO2 standard curve.
Enzyme-linked immunosorbent assay
RAW 264.7 macrophages were seeded overnight (200,000 cells per well) in 24-well plates (BD Falcon). Cells were then stimulated with lipopolysaccharide (100 ng/mL) for 24 hours, in the presence of increasing concentrations of the extract. Supernatants were collected and subjected to enzyme-linked immunosorbent assay (ELISA) to quantify granulocyte macrophage–colony-stimulating factor (GM-CSF) and tumor necrosis factor (TNF)-α release. Quantikine™ Mouse ELISA kits (R&D Systems) were used, and the protocols provided by the manufacturers were carefully followed. All samples were analyzed in triplicate, and data were acquired by using an automated Varioskan plate reader.
Western blots
RAW 264.7 cells were seeded in a Petri dish at a density of 2×107 cells per dish, then stimulated and exposed to extracts as described above. After 24-hour incubation at 37°C and 5% CO2, cells were harvested and homogenized with a lysis buffer. Protein concentrations were determined by using the Bradford method, with bovine serum albumin as standard. Fifteen micrograms of proteins was loaded on 10% sodium dodecyl sulfate–polyacrylamide gels. 17 After electrophoresis, proteins were transferred to a polyvinylidene fluoride membrane, blocked with 5% dry milk for 1 hour, then probed with antibodies as follows: Actin was detected with 1:150 rabbit primary antibodies (Sigma A2066) and 1:5000 anti-rabbit secondary antibodies (Amersham Na934). Cyclooxygenase-2 (COX-2) was revealed with 1:5000 goat primary antibodies (Santa Cruz Se1745) and 1:7000 anti-goat secondary antibodies (Santa Cruz Sc2352). Inducible nitric oxide synthase (iNOS) was detected with 1:10000 rabbit primary antibodies (Sigma N7782) and 1:7000 anti-rabbit secondary antibodies (Amersham Na934). All antibodies were diluted in 5% milk with Tris-buffered saline and Tween 20 and applied for 1 hour each at room temperature. Images were acquired with a Chemidoc XRS, and densitometry analysis performed with Quantity One software, version 4.5.2.
Statistical analysis
Student t-tests were used to determine statistical significance. The results were considered significantly different when P value is .05 or less (P≤.05). SigmaStat software version 3.5 was used for statistical analysis.
Results and Discussion
Antioxidant activity evaluation of B. schreberi leaf extract and isolation of the main compounds
Three B. schreberi leaf extracts were obtained by sequential extraction with dichloromethane, methanol, and water. The antioxidant potential of each extract was assessed by using the ORAC assay. As shown in Table 1, the dichlomethane extract was slightly active, with a mean±standard deviation ORAC value of 0.8±0.4 μmol TE/mg, whereas the methanol and water extracts were strongly active, with ORAC values of 7±2 and 5.1±0.5 μmol TE/mg, respectively. The main compounds of the methanolic and water extracts were isolated to identify the compounds responsible for their antioxidant activities (Fig. 1). The methanolic extract was treated with n-BuOH-saturated H2O, the n-BuOH fraction was repeatedly chromatographed on silica gel columns, and 8 fractions were collected. Major compound 1 was found in fraction 4 and was isolated by HPLC for identification. The water extract was extracted with ethyl acetate and subjected to silica gel column chromatography. Seven fractions were collected; main compound 2 was found in fraction 2 and was isolated by HPLC for identification.
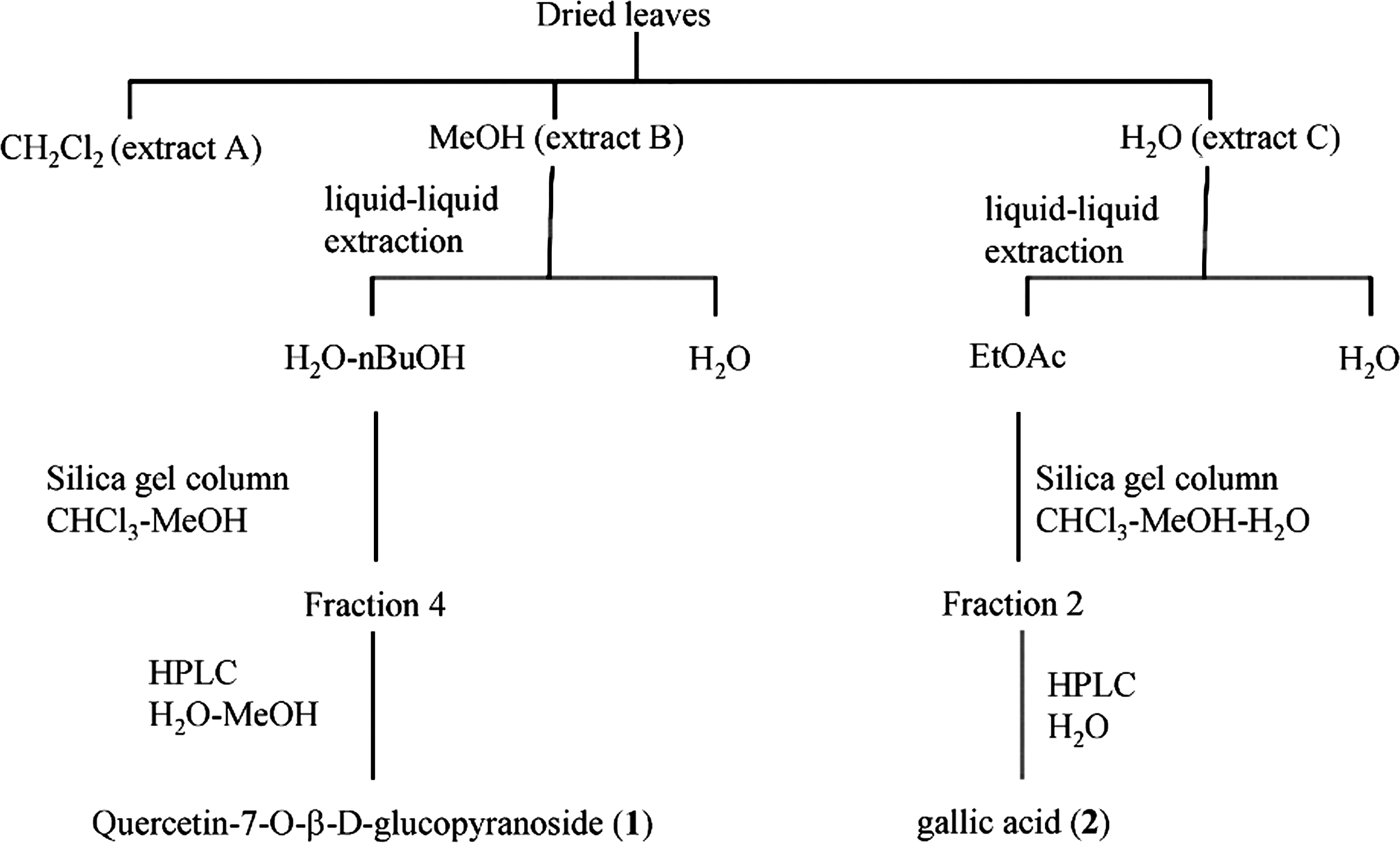
Isolation paths of major compounds from Brasenia schreberi leaf extracts. EtOAc, ethyl acetate.
ORAC results for extracts are expressed as μmol Trolox equivalents/mg). Quercetin, quercetin 7-O-β-D-glucopyranoside, and gallic acid are expressed as μmol Trolox equivalents/μmol).
ORAC, oxygen radical absorbance capacity; SD, standard deviation.
Identification of the main compounds from the leaves of B. schreberi
Compound 1 exhibited a peak at m/z=487.0844 in the HR ESI-MS analysis, corresponding to a molecular formula of C21H20O12Na, which was supported by 13C NMR data (see Materials and Methods section). The APCI-MS spectrum of 1 showed peaks at m/z 301 (loss of 162), suggesting the presence of a hexose bonded to an aglycone moiety. Configuration of the sugar moiety was determined to be β on the basis of the coupling constant of the anomeric proton (J1″,2″ =7.6 Hz). In addition, the anomeric proton showed a correlation in the HMBC spectrum with δC 163.7 (C-7). Thus, according to these findings and all the spectroscopic data (see Materials and Methods section), the structure of compound 1 (Fig. 2) was determined to be quercetin-7-O-β-D-glucopyranoside, which was further confirmed by comparison with data from the literature. 18,19 Compound 2 exhibited a blue-violet color on TLC with use of the NP/PEG reagent, a finding that is characteristic of phenolic compounds. 12 The molecular formula was deduced as being C7H6O5 according to HR ESI-MS results (see Materials and Methods section). Finally, comparison of APCI-MS and NMR data with compound standard confirmed that compound 2 was gallic acid (Fig. 2). To our knowledge, quercetin-7-O-β-D-glucopyranoside and gallic acid were identified for the first time in B. schreberi leaves. However, quercetin-7-O-β-D-glucopyranoside has previously been isolated from Chrysanthemum segetum, Asclepias syriaca, and Carthamus tinctrius, and gallic acid is a widespread phenolic compound. 20 –23
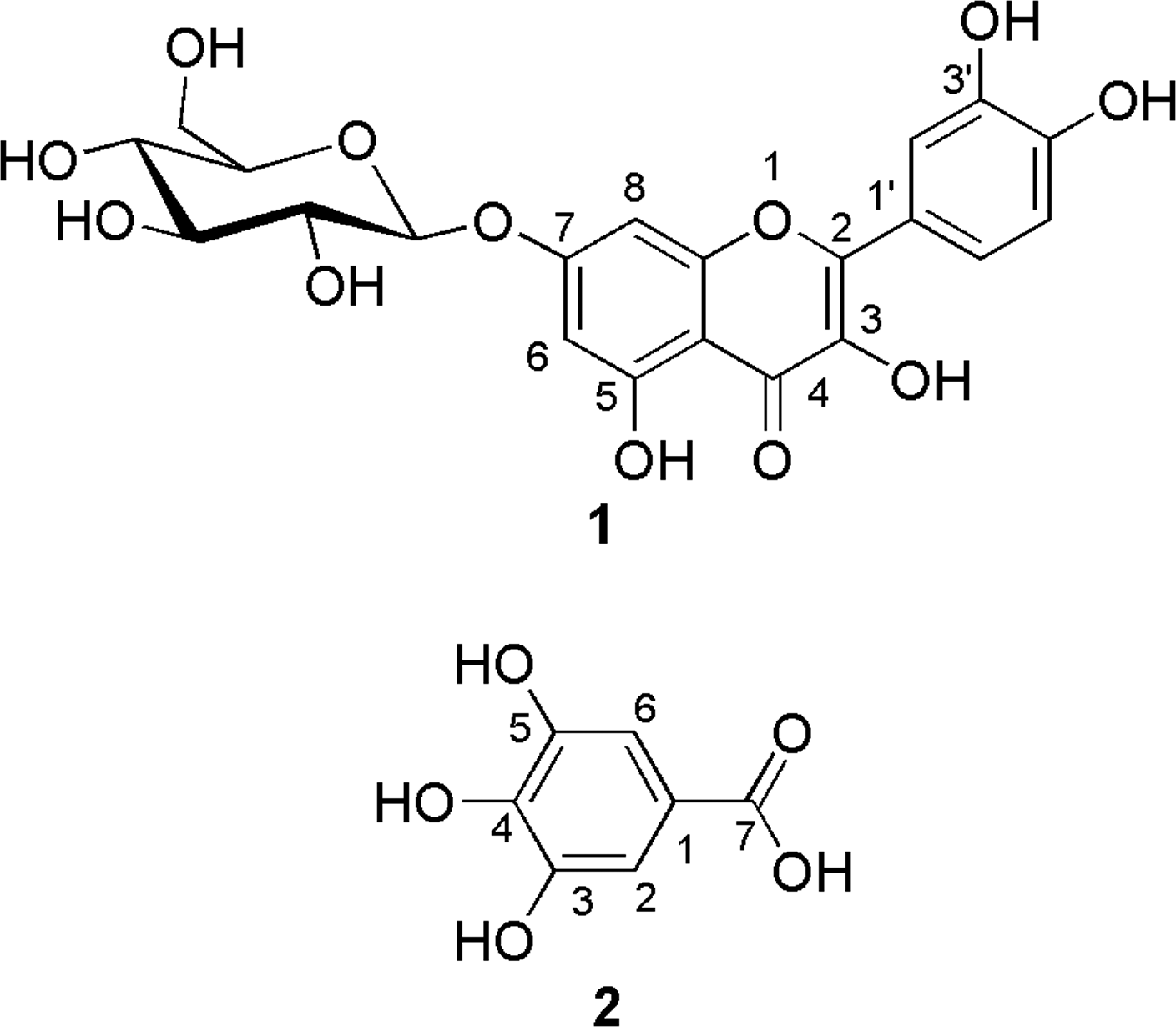
Molecular structures of quercetin-7-O-β-D-glucopyranoside (1) and gallic acid (2) isolated from Brasenia schreberi.
Antioxidant activity and cytotoxicity evaluation of quercetin-7-O-β-D-glucopyranoside (1)
The antioxidant activities of quercetin-7-O-β-D-glucopyranoside and gallic acid were assessed by using the in vitro ORAC assay. The results, displayed in Table 1, indicate that quercetin-7-O-β-D-glucopyranoside and gallic acid are antioxidants, with ORAC values of 18±4 and 4±1 μmol/TE μmol, respectively. Quercetin-7-O-β-D-glucopyranoside was found more active than quercetin, a strong antioxidant possessing an ORAC value of 10±1 μmol TE/μmol. These results suggest that quercetin-7-O-β-D-glucopyranoside and gallic acid are partly responsible for the antioxidant activity of the methanolic and water extracts, respectively.
The antioxidant activity of quercetin-7-O-β-D-glucopyranoside and gallic acid using in vitro assays was previously reported. 24,25 To support the results obtained in vitro, the antioxidant activity of quercetin-7-O-β-D-glucopyranoside was also assessed by using an in vitro cell-based assay. The results (Fig. 3) show that 0.25 and 1 μg/mL of quercetin-7-O-β-D-glucopyranoside inhibited the tert-butyl hydroperoxide–induced oxidation of DCFH by roughly 52% and 68%, respectively. In comparison, Trolox (0.25 μg/mL), a water-soluble vitamin E derivative, and quercetin (0.25 μg/mL), inhibited the tert-butyl hydroperoxide–induced oxidation of DCFH by about 51% and 80%, respectively. However, although quercetin was cytotoxic for L-929 cells, no such cytotoxicity was observed for quercetin-7-O-β-D-glucopyranoside.
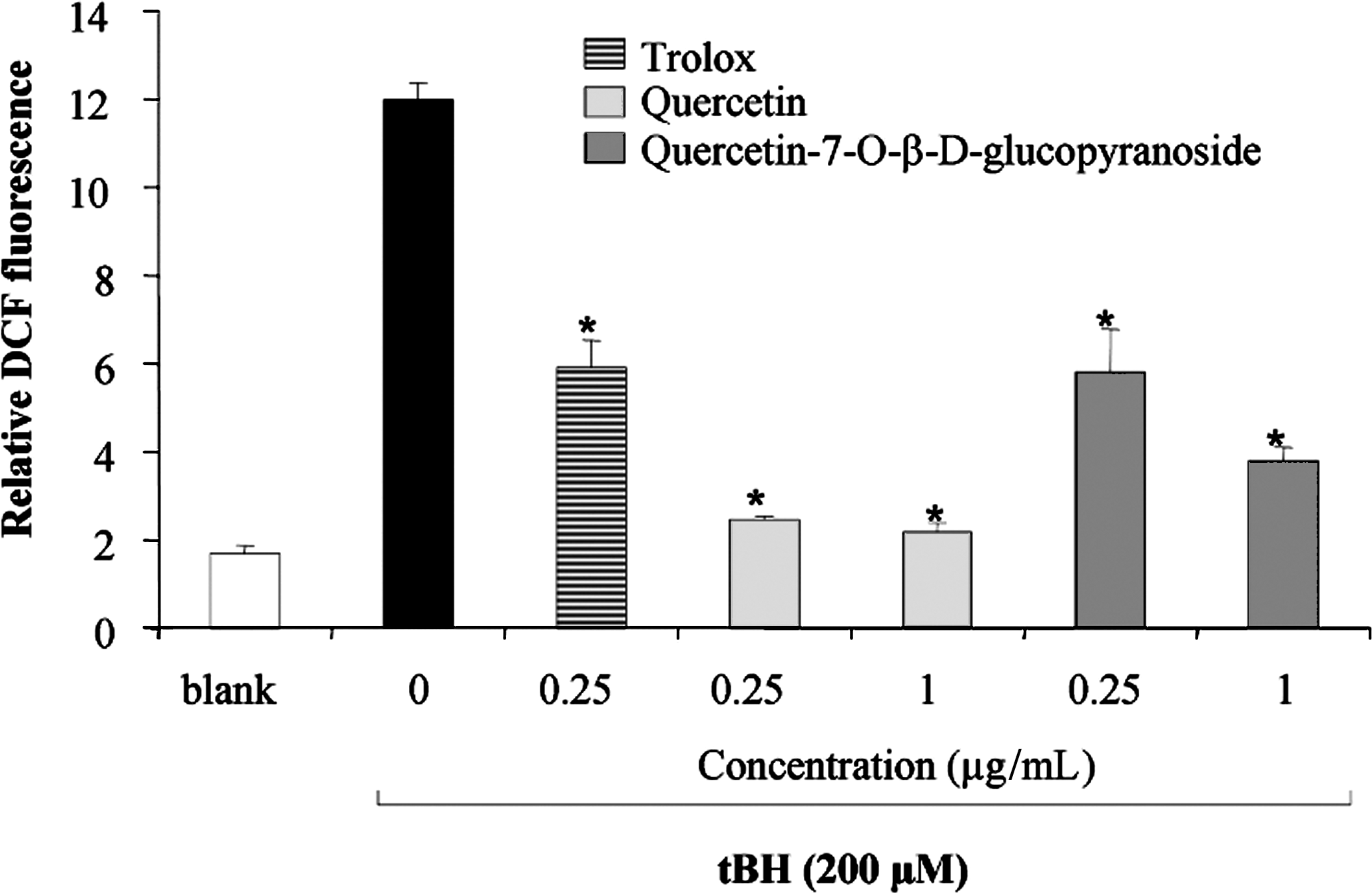
Effect of quercetin and quercetin-7-O-β-D-glucopyranoside on reactive oxygen species production induced by tert-butyl hydroperoxide in L-929 cell lines. Trolox (0.25 μg/mL) was used as standard. Values are reported as mean±standard deviation and are representative of 3 different determinations in triplicate. *Significantly different from cells treated with tert-butyl hydroperoxide, 200 μM only; P≤.05, Student t-test. DCF, dichlorofluorescein; tBH, tert-butyl hydroperoxide.
The cytotoxic activities of quercetin and quercetin-7-O-β-D-glucopyranoside toward normal human skin fibroblasts (WS-1) were also assessed. The cells were incubated for 48 hours in the presence or absence of growing concentrations (doses ranging from 1 to 36 μg/mL) of quercetin-7-O-β-D-glucopyranoside or quercetin. The results indicate that quercetin-7-O-β-D-glucopyranoside was not cytotoxic toward WS-1, with 95% cell survival at 36 μg/mL (Fig. 4). In contrast, quercetin was cytotoxic in a dose-dependent manner; cell growth was inhibited by 50% at 6.6±0.4 μg/mL.
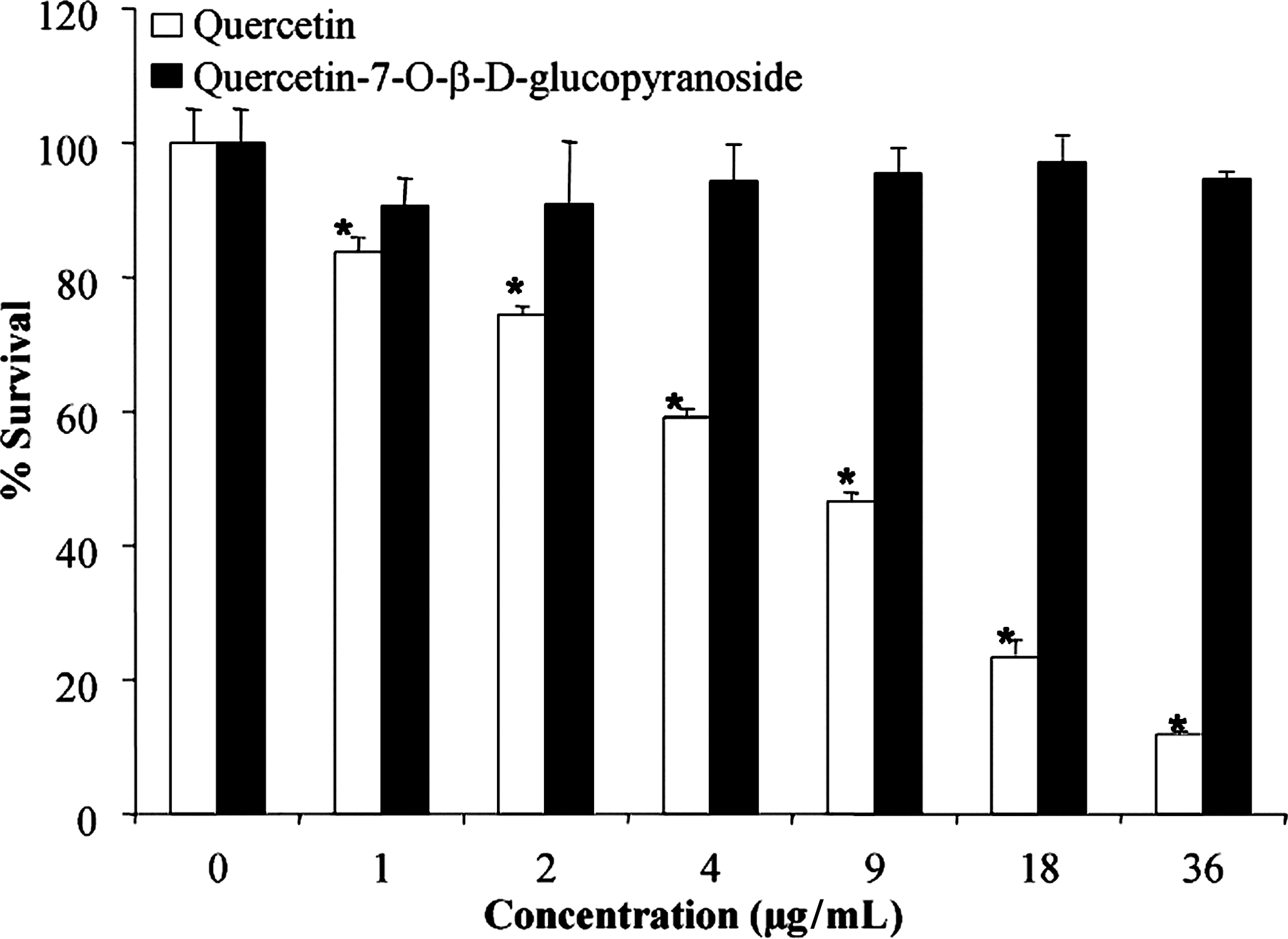
Cytotoxicity of quercetin and quercetin-7-O-β-D-glucopyranoside on human fibroblast cell line WS-1. Values are reported as mean±standard deviation of and are representative of 3 different determinations in triplicate. *Significantly different from control; P≤.05, Student t-test.
Quercetin has previously been reported as cytotoxic toward normal cultured human cells 26 and as mutagenic and genotoxic in vitro. 27,28 The cytotoxicity of quercetin could be due to an increase of intracellular reactive oxygen species. Indeed, quercetin may act as a pro-oxidant after the intracellular metabolic activation of quercetin to o-quinone. 29 Long-term administration of quercetin in rats also showed a pro-oxidant effect. 30 Of note, the presence of glucose in position 7 of quercetin inhibits cytotoxicity. The metabolism of quercetin glucosides and their bioavailability in human plasma is not clear. Paganga and Rice-Evans measured quercetin glucosides in human plasma and suggested that it is absorbed intact by the intestine. 31 On the other hand, Olthof et al. reported that quercetin aglycone is present in human plasma after ingestion of quercetin glycosides, indicating that sugar is cleaved by glucosidase in the intestine. 32 In contrast, Sesink et al. found no intact quercetin glucosides and only trace amounts of aglycone in human plasma after consumption of quercetin glucosides. Indeed, these authors show that quercetin glucuronides are major metabolites in plasma. 33 Additional studies are needed to verify these contradictory observations.
Anti-inflammatory activity evaluation of quercetin-7-O-β-D-glucopyranoside (1) on murine RAW 264.7 macrophages
A variety of in vitro and in vivo experiments have shown that several flavonoids possess anti-inflammatory activities. 34 Moreover, flavonoids have been reported to possess beneficial effects on cardiovascular and chronic inflammatory diseases associated with NO. 35 Jung and Sung showed that quercetin treatment of lipopolysaccharide-stimulated murine RAW 264.7 macrophages significantly reduced nitrite release, indicating that some dietary antioxidants possess significant anti-inflammatory activities. 36 However, these authors did not investigate cytotoxicity of quercetin toward RAW 264.7 cells. In our study, we assessed the cytotoxic effects of quercetin and quercetin-7-O-β-D-glucopyranoside on growing RAW 264.7 cells. The results, shown in Figure 5A, reveal that quercetin (4 μg/mL) was cytotoxic, with a 66% cell survival rate. In contrast, quercetin-7-O-β-D-glucopyranoside was not significantly cytotoxic toward RAW 264.7 cell lines at doses ranging from 1 to 16 μg/mL. The anti-inflammatory activity of quercetin-7-O-β-D-glucopyranoside was evaluated on lipopolysaccharide-stimulated RAW 264.7 macrophages as described in the Materials and Methods section. L-NAME, an NO synthase inhibitor that prevents the overproduction of NO in lipopolysaccharide-stimulated RAW 264.7 cells, was used as a positive control. 37 The results (Fig. 5B) show that quercetin-7-O-β-D-glucopyranoside significantly inhibited overproduction of NO in a dose-dependent manner, with an inhibition of 40%±2% at 16 μg/mL. In comparison, the positive control L-NAME prevented NO release by 44%±2% at 62.5 μg/mL.

Effect of quercetin-7-O-β-D-glucopyranoside on (
NO is generated via overexpression of iNOS, which is stimulated by lipopolysaccharide. 38 Moreover, lipopolysaccharide induces COX-2 in RAW264.7 cells. 39 COX-2 is implicated in the biosynthesis of prostaglandin as prostaglandin E2, which is responsible for edema and pain. 40 The effect of quercetin-7-O-β-D-glucopyranoside on iNOS and COX-2 overexpression was evaluated by using Western blot (Fig. 6). As expected, lipopolysaccharide (100 μg/mL) induces overexpression of iNOS and COX-2 in comparison with untreated cells. Interestingly, quercetin-7-O-β-D-glucopyranoside inhibited about 95% overexpression of iNOS induced by lipopolysaccharide at doses of 15 and 30 μg/mL. Moreover, the results show that 15 and 30 μg/mL of quercetin-7-O-β-D-glucopyranoside inhibited, respectively, about 50% and 100% overexpression of COX-2 induced by lipopolysaccharide.
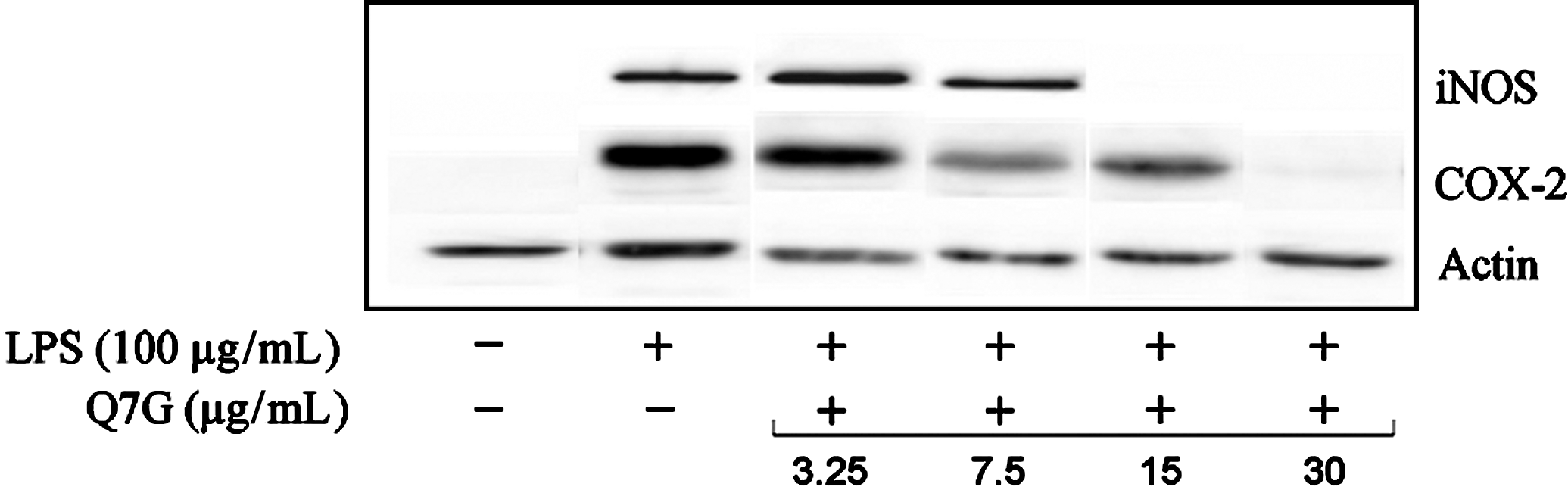
Effects of quercetin-7-O-β-D-glucopyranoside (Q7G) on iNOS and COX-2 expression in LPS-stimulated RAW 264.7 macrophages, analyzed by Western blotting. Actin was used as loading control. COX, cyclooxygenase; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide.
Lipopolysaccharide-stimulated RAW 264.7 cells are also known to produce various pro-inflammatory cytokines, such as GM-CSF and TNF-α. The effects of quercetin-7-O-β-D-glucopyranoside on GM-CSF and TNF-α overproduction were examined after 24 hours of treatment in the presence of lipopolysaccharide. Figure 7 shows that quercetin-7-O-β-D-glucopyranoside significantly inhibits lipopolysaccharide-induced GM-CSF production in a concentration-dependent manner, with an inhibition of 53%±6% at 4 μg/mL. However, quercetin-7-O-β-D-glucopyranoside did not inhibit TNF-α production induced by lipopolysaccharide.

Effect of quercetin-7-O-β-D-glucopyranoside on (
In conclusion, 2 phenolic compounds, quercetin-7-O-D-β-glucopyranoside (1) and gallic acid (2), were identified for what we believe to be the first time in B. schreberi leaves, and both compounds exhibit strong antioxidant activities. Quercetin-7-O-β-D-glucopyranoside possesses interesting anti-inflammatory activity, inhibiting NO release, iNOS and COX-2 expression, and GM-CSF overproduction. Unlike quercetin, it was not found to be cytotoxic in vitro.
Footnotes
Acknowledgments
The authors wish to kindly thank Dr. F-X. Garneau for his precious help and suggestions. The authors also thank Patrick Nadeau for plant identification. This work was supported by the Fondation de l'Université du Québec à Chicoutimi and by Action Concertée FQRNT - Fonds de la recherche forestière du Saguenay - Lac-Saint-Jean.
Author Disclosure Statement
No competing financial interests exist.