
Abstract
Adrenocortical carcinomas are rare but present with extremely poor prognosis. One of the approaches to control cancer progression and reduce cancer risk is prevention through diet. Bitter melon is widely consumed as a vegetable and especially as a traditional medicine in many countries. In this study, we have used human and mouse adrenocortical cancer cells as an in vitro model to assess the efficacy of bitter melon extract (BME) as an anticancer agent. The protein concentrations of BME and other extracts were measured before use. First, BME treatment of adrenocortical cancer cells resulted in a significantly dose-dependent decrease in cell proliferation. However, we did not observe an antiproliferative effect in adrenocortical cancer cells treated with extracts from blueberry, zucchini, and acorn squash. Second, apoptosis of adrenocortical cancer cells was accompanied by increased caspase-3 activation and poly(ADP-ribose) polymerase cleavage. BME treatment enhanced cellular tumor antigen p53, cyclin-dependent kinase inhibitor 1A (also called p21), and cyclic AMP–dependent transcription factor-3 levels and inhibited G1/S-specific cyclin D1, D2, and D3, and mitogen-activated protein kinase 8 (also called Janus kinase) expression, suggesting an additional mechanism involving cell cycle regulation and cell survival. Third, BME treatment decreased the key proteins involved in steroidogenesis in adrenocortical cancer cells. BME treatment decreased the level of phosphorylation of cyclin-dependent kinase 7, which is required, at least in part, for steroidogenic factor 1 activation. Finally, we observed that BME treatment significantly reduced the level of insulin-like growth factor 1 receptor and its downstream signaling pathway as evidenced by lower levels of phosphorylated RAC-α serine/threonine-protein kinase. Taken together, these data illustrate the inhibitory effect of bitter melon on cell proliferation of adrenocortical cancer through modulation of diverse mechanisms.
Introduction
A
Currently, approximately 45% of modern drugs are derived from natural sources such as plants. 6 Bitter melon (Momordica charantia) is a tropical and subtropical vine that produces fruits that can be used as traditional herbal medicine to treat diabetes and insulin resistance in East Asia, India, and Central America. 7 –9 Crude bitter melon extract (BME) and several active ingredients from bitter melon have been shown to improve blood glucose, stimulate insulin secretion, reduce hepatic triglyceride production, and enhance leptin secretion in cell lines and animal models. 8 –12 In the last decades, extensive studies have demonstrated that bitter melon produces ribosome-inactivating proteins that possess antitumor and antiviral activities. 13 –15 Several lines of evidence have shown that BME preparations (from leaves, seeds, or fruits) block prostate, breast, and colon cancer growth by several different mechanisms, including oxidation-dependent mechanism, apoptosis, and/or cell cycle regulation. 13 –23 Therefore, the use of bitter melon as a functional food appears to have a health benefit.
The objective of this study was to examine BME efficacy against adrenocortical cancer cells. We chose crude BME for this study because strong evidence from blueberry studies has suggested that whole extract exhibited higher antiproliferative activity than fractions or compounds. 24 Our results show that treatment of adrenocortical cancer cells with BME induces cell death and impaired cell cycle progression, reduces steroidogenesis, and inhibits the IGF1R signaling pathway, suggesting an important role of BME on prevention of adrenocortical cancers.
Materials and Methods
Reagents
All cell culture reagents were purchased from Invitrogen (Carlsbad, CA, USA). Luciferase activity was measured using the Dual Luciferase Assay System (Promega, Madison, WI, USA). Antibodies against cellular tumor antigen p53 (TP53), cyclic AMP–dependent transcription factor-3 (ATF3), G1/S-specific cyclin D2 (CCND2), melanocortin type 2 receptor (MC2R), tubulin, cyclin-dependent kinase (CDK) inhibitor 1A (CDKN1A) (also called p21), CDK7, β-catenin (CTNNB1), IGF1β, histone deacetylase (HDAC) 1, and HDAC2 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Antibodies against G1/S-specific cyclin D1 (CCND1), G1/S-specific cyclin D3 (CCND3), RAC-α serine/threonine-protein kinase (AKT) (total), phospho-AKT (Ser473), extracellular signal-regulated kinase (ERK) 1/2 (total), phospho-ERK1/2, caspase (CASP)-3, cleaved CASP9, and cleaved poly(ADP-ribose) polymerase (PARP) were purchased from Cell Signaling Technology Inc. (Danvers, MA, USA). Antibodies against steroidogenic factor 1 (NR5A1) were purchased from Upstate Biochemistry Inc. (Charlottesville, VA, USA). Antibodies against β-actin were purchased from Sigma (St. Louis, MO, USA). Antibodies against steroidogenic acute regulatory protein (STAR) and phospho-CDK7 were purchased from Abgent (San Diego, CA, USA).
DNA constructs
The murine Mc2r promoter (2,000 bp) was constructed as previously described. 25 The murine Nr5a1 promoter (700 bp) was polymerase chain reaction–amplified by using forward primer 5′-ACGAGGTACCTCTAGACAAAAAAC-3′ and reverse primer 5′-GCCAAGCTTAATTCTCAGCCGG-3′, digested with KpnI and HindIII, and then ligated into the KpnI and HindIII sites of pGL3 to create Nr5a1 promoter luciferase plasmid. The human CCND1 promoter (700 bp) was polymerase chain reaction–amplified by using forward primer 5′-ACGAGGTACCTAAAAAAAATGAGTCAG-3′ and reverse primer 5′-GCCAAGCTTCCCCGCTGCAGCCTTTC -3′, digested with KpnI and HindIII, and then ligated into the KpnI and HindIII sites of pGL3 to create CCND1 promoter luciferase plasmid. Mouse Star luciferase reporter plasmid, human TP53 luciferase reporter plasmid, and human mitogen-activated protein kinase (MAPK) 8 (c-Jun N-terminal kinase 1 [JNK1]) luciferase reporter plasmid were kindly provided by Dr. Kenneth Escudero (Texas A&M University, Kingsville, TX, USA), Dr. Junga Kawauchi, and Drs. R.H. Costa and P. Raychandhuri (University of Illinois, Chicago, IL, USA), respectively. Mouse Igf2 luciferase reporter plasmid was kindly provided by Dr. A. Murrell (Cambridge Research Institute, Cambridge, United Kingdom). All constructs were verified by nucleotide sequencing.
Preparation of BME, blueberry extract, zucchini extract, and acorn squash extract
Whole fresh Chinese variety young bitter melons (green and raw) from a local farmer's market (Savannah, GA, USA) were washed and deseeded. BME was extracted using a household juicer and centrifuged (Allegra X-22R, Beckman Coulter, Brea, CA, USA) at 4000 rpm at 4°C for 30 minutes. The supernatant BME was filtered (pore size, 0.22 μm; Millipore, Milford, MA, USA) and stored in aliquots at −80°C until further analysis. Each aliquot was frozen and thawed only once to prevent protein and lipid degradation. Whole fresh blueberries, zucchini, and acorn squash were also purchased from the same farmer's market. Blueberry extract, zucchini extract, and acorn squash extract were prepared similarly as described for BME preparation. The protein concentration of each crude extract was determined using the Bradford protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA): BME, 11.6±1.2 mg/mL; blueberry extract, 11.6±1.8 mg/mL; zucchini extract, 9.6±1.3 mg/mL; and acorn squash extract, 15.5±1.6 mg/mL.
Cell culture
NCI H295R (H295R) cells, purchased from the American Type Culture Collection (Manassas, VA, USA), were maintained in Dulbecco's modified Eagle's medium:F12 medium supplemented with 2.5% Nu-serum (BD Biosciences, Sparks, MD, USA), ITS+ Premix (BD Biosciences), and antibiotics in humidified air containing 5% CO2, at 37°C. Y1 cells, described previously, 25 were maintained in Dulbecco's modified Eagle's medium supplemented with 7.5% horse serum, 2.5% fetal bovine serum, and antibiotics in humidified air containing 5% CO2, at 37°C.
Primary cell culture of rat adrenal cells
Adrenal glands were obtained from normal adult rats (male Sprague–Dawley rats weighing approximately 200–250 g). All animal studies have been approved by the Mercer University Animal User and Care Committee. Primary cell cultures of rat adrenal cells were established from adrenal glands of three normal rats within 1 hour after surgery. Normal adrenal glands were freed of fat, minced, and incubated for 60 minutes at 37°C in phosphate-buffered saline containing 2 mg/mL collagenase (Sigma). To facilitate dispersion, tissue was minced with a Pasteur pipette with a fine heat-polished tip, and the cell suspension was filtered through a cell strainer (pore size mesh, 80 μm; Sigma-Aldrich Corp., St. Louis) and centrifuged for 10 minutes at 200 g. The pellet was resuspended in a culture medium consisting of a 1:1 (vol/vol) mixture of Dulbecco's modified Eagle's medium:F12 medium with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin, enriched with a mixture of ITS+ Premix solution. Isolated cells were cultured in humidified air containing 5% CO2, at 37°C.
Cell viability assay
Cells were treated with 100 μL of medium plus water or extract and incubated for 54 hours. Extract was tested at 0%, 0.5%, 1%, 2%, 4%, and 8% (vol/vol) concentrations. At the end of the extract exposure duration, cell viability was measured using the CellTiter-Glo® Luminescent Cell Viability Assay according to the manufacturer's instructions (Promega). All plates had control wells containing medium without cells to obtain a value for background luminescence, which was subtracted from the test sample readings. All experiments were performed three times in triplicate.
Western blotting
Y1 and H295R cells were treated with (1% or 2% [vol/vol]) or without BME, and cell lysates were prepared 36 hours after treatment in radioimmunoprecipitation assay (RIPA) buffer. Protein lysates were allowed to rotate at 4°C for 30 minutes, and protein contents of the high-speed supernatant were determined using the Bradford protein assay (Bio-Rad). Equivalent quantities of protein (20–45 μg) were resolved on 7.5–10% polyacrylamide–sodium dodecyl sulfate gels, transferred to a nitrocellulose membrane (Bio-Rad), and immunoblotted with specific antibodies. Results were visualized using the SuperSignal West Dura Extended Duration Substrate kit (Pierce Chemical Co., Rockford, IL, USA). Band intensity was quantified with the ImageJ program (National Institutes of Health, Bethesda, MD, USA).
Reporter gene assay
Y1 and H295R cells were transiently transfected with luciferase expression construct using FuGENE® HD Transfection Reagent (Roche, Indianapolis, IN, USA) according to the manufacturer's protocol. Six hours posttransfection, cells were treated with various concentrations of BME for 24 hours. Cell lysates were collected, and luciferase activity was measured and normalized with Renilla activity. All experiments were performed three times in triplicate.
Statistical analysis
Statistical analyses were performed using Student's t test or a one-way analysis of variance when more than two groups were compared. After the analysis of variance, the post hoc multiple comparisons were performed by using Tukey's Honestly Significant Difference test to determine the statistical difference from each other among subgroups. For each test, P values of < .05 and < .001 were considered significant and very significant, respectively.
Results
BME treatment induces adrenocortical cancer cell death
BME was tested in human H295R and mouse Y1 adrenocortical cell lines and normal rat adrenal cells. Cell viability was measured after 54 hours. As shown in Figure 1A, we observed approximately 50% cell death in H295R and Y1 cells treated with 2% (vol/vol) BME. On the other hand, primary adrenal cells from rats did not display any antiproliferation with 2% BME. When 4% and 8% (vol/vol) BME induced >90% cell death in H295R and Y1 cells, primary adrenal cells displayed less than 50% cytotoxicity even at 8% BME treatment. This result indicates that the antiproliferative effect of BME was most effective in cancer cells. Recent studies have shown that blueberry extract inhibited growth of breast cancer cells. Thus, we next compared the antiproliferative effect of BME and blueberry extract on Y1 and H295R cells. As shown in Figure 1B and C, we observed that BME, but not blueberry extract, exhibited a dose-dependent antiproliferation effect on adrenocortical cancer cells. As expected, extracts from acorn squash and zucchini, two vegetables from the same Cucurbitaceae family as bitter melon, did not induce cell death in adrenocortical cancer cells.
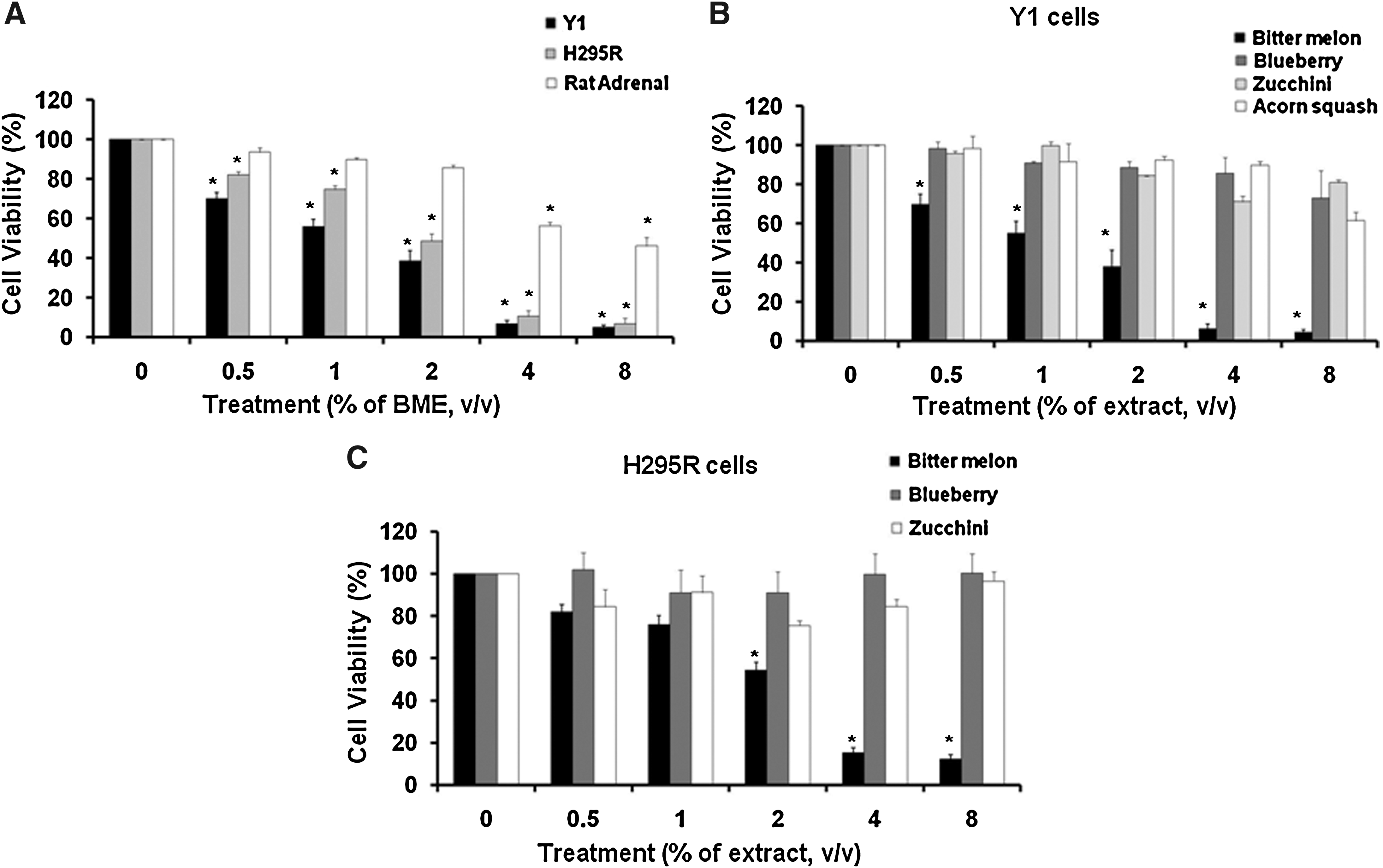
Bitter melon extract (BME) induces adrenocortical cancer cell death.
BME induces CASP activation and PARP cleavage in adrenocortical cancer cells
The 116-kDa nuclear PARP has shown to be involved in DNA repair in response to environmental stress and stimuli. PARP is one of the main target substrates of CASP3 in vivo. Cleavage of PARP serves as a marker of cells undergoing apoptosis. To investigate the effect of BME on apoptosis, PARP cleavage was determined by immunoblotting in Y1 and H295R cells treated with different concentrations of BME. As shown in Figure 2, both cell lines treated with BME showed induction of PARP cleavage and displayed activation of CASP3. Moreover, CASP9, which is involved in the activation of the CASP proteins responsible for apoptosis execution, was activated in H295R cells treated with BME (Fig. 2B). These results suggest that BME treatment triggers CASP cascades eventually leading to activation of apoptosis in adrenocortical cancer cells.

BME induces caspase (CASP) activation and poly(ADP-ribose) polymerase (PARP) cleavage in adrenocortical cancer cells.
Cell cycle regulatory proteins were modulated following BME treatment
TP53 protein acts as a major player in cellular response to DNA damage and other genomic stresses. Activation of TP53 can lead to cell cycle arrest and apoptosis. CDKN1A (also called p21) protein serves as an inhibitor of cell cycle progression, by forming complexes with cyclins and CDKs. ATF3, a stress sensor, is rapidly induced by a wide range of stresses, including DNA-damaging agents, ultraviolet radiation, and cytotoxicity. Therefore, we next examined these key cell cycle regulatory proteins in adrenocortical cancer cells following BME treatment. Y1 and H295R cells were treated with 1% (vol/vol) and 2% (vol/vol) BME for 36 hours, followed by immunoblotting analysis. As shown in Figure 3A and B, BME-treated adrenocortical cell lysates displayed up-regulation of TP53 and CDKN1A levels and inhibition of the CCND family of proteins. ATF3 levels were increased in 1% but decreased in 2% BME-treated cell lysates, supporting ATF3's role as a stress sensor. These results were further examined by reporter gene assay. BME treatment significantly reduced CCND1 transcriptional activity in H295R cells (Fig. 3C). On the other hand, TP53 transcriptional activity was significantly enhanced by 1% (vol/vol) BME treatment in H295R cells. We also found that BME treatment significantly reduced MAPK8 (also called JNK1) transcriptional activity in H295R cells. These results suggest that BME uses several signaling pathways to induce cell death and impair cell cycle regulation of adrenocortical cancer cells.
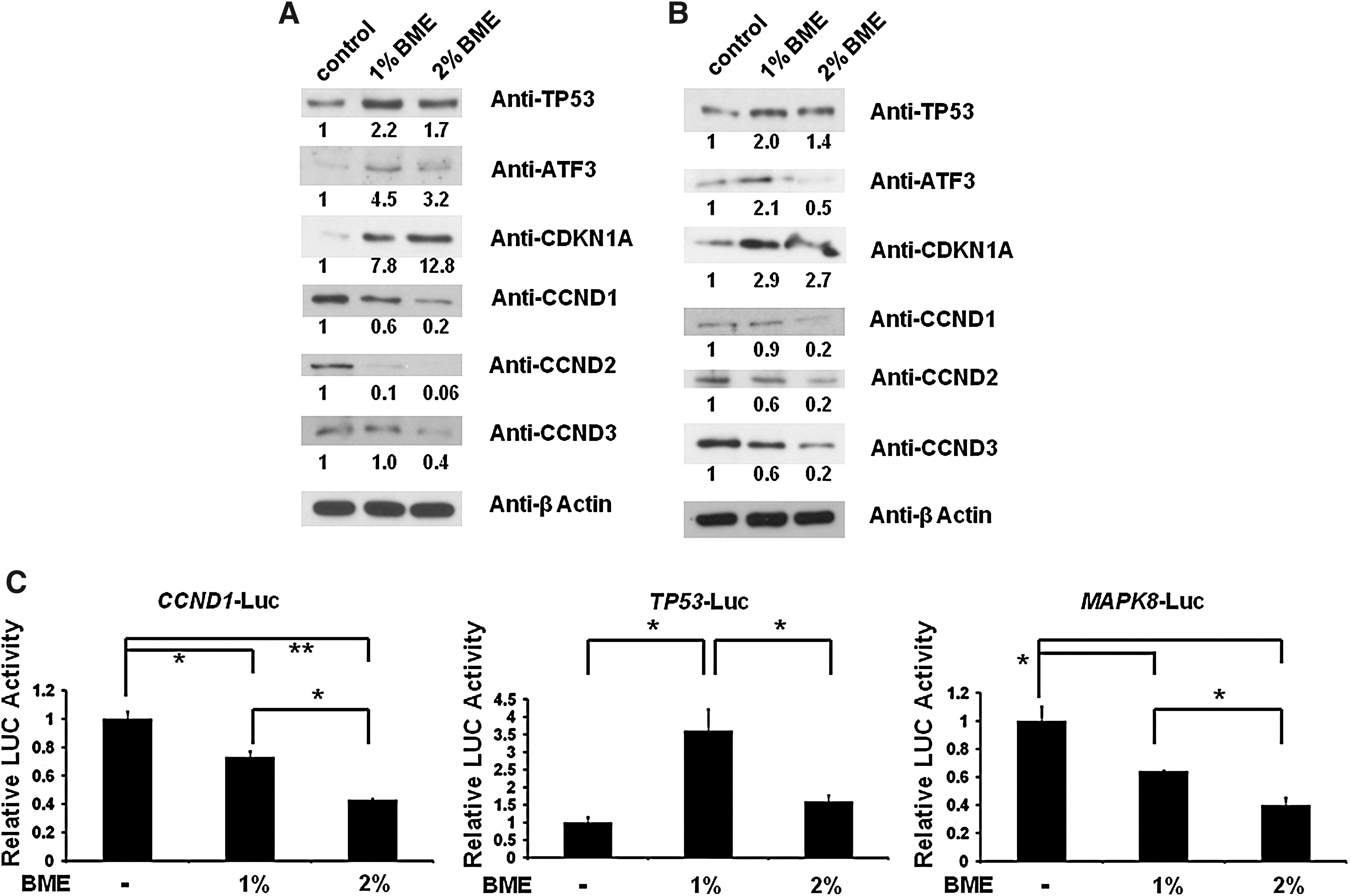
Cell cycle regulatory proteins were modulated following BME treatment in adrenocortical cancer cells.
BME suppresses key regulatory proteins involved in steroidogenesis
Because of the importance of the steroidogenic signaling pathway in adrenocortical cells, we next determined the effect of BME on the key regulatory proteins (NR5A1, MC2R, and STAR) involved in adrenocortical steroidogenesis. As shown in Figure 4A and 4B, BME treatment in both Y1 and H295R cells significantly reduced NR5A1, MC2R, and STAR protein levels. These results were further supported by the reporter gene assay in Y1 cells (Fig. 4C). We have previously shown that NR5A1 is activated, at least in part, by CDK7. We then investigated whether BME treatment alters CDK7 activity. As expected, phosphorylation of CDK7 protein (activated form) levels were reduced by BME treatment in both Y1 and H295R cells. However, total CDK7 protein levels were unaffected. Because of constitutive activation of CTNNB1 (also called β-catenin), the most frequent alteration in benign and malignant adrenocortical tumors, we further investigated whether BME treatment alter CTNNB1 protein levels in Y1 cells (Fig. 4A). To our surprise, CTNNB1 levels were not altered even at 2% (vol/vol) BME treatment. Similarly, BME treatment did not alter HDAC1 and HDAC2 protein levels in Y1 cells. These results indicate that the BME treatment in adrenocortical cancer cells affects their normal regulation of steroidogenesis.
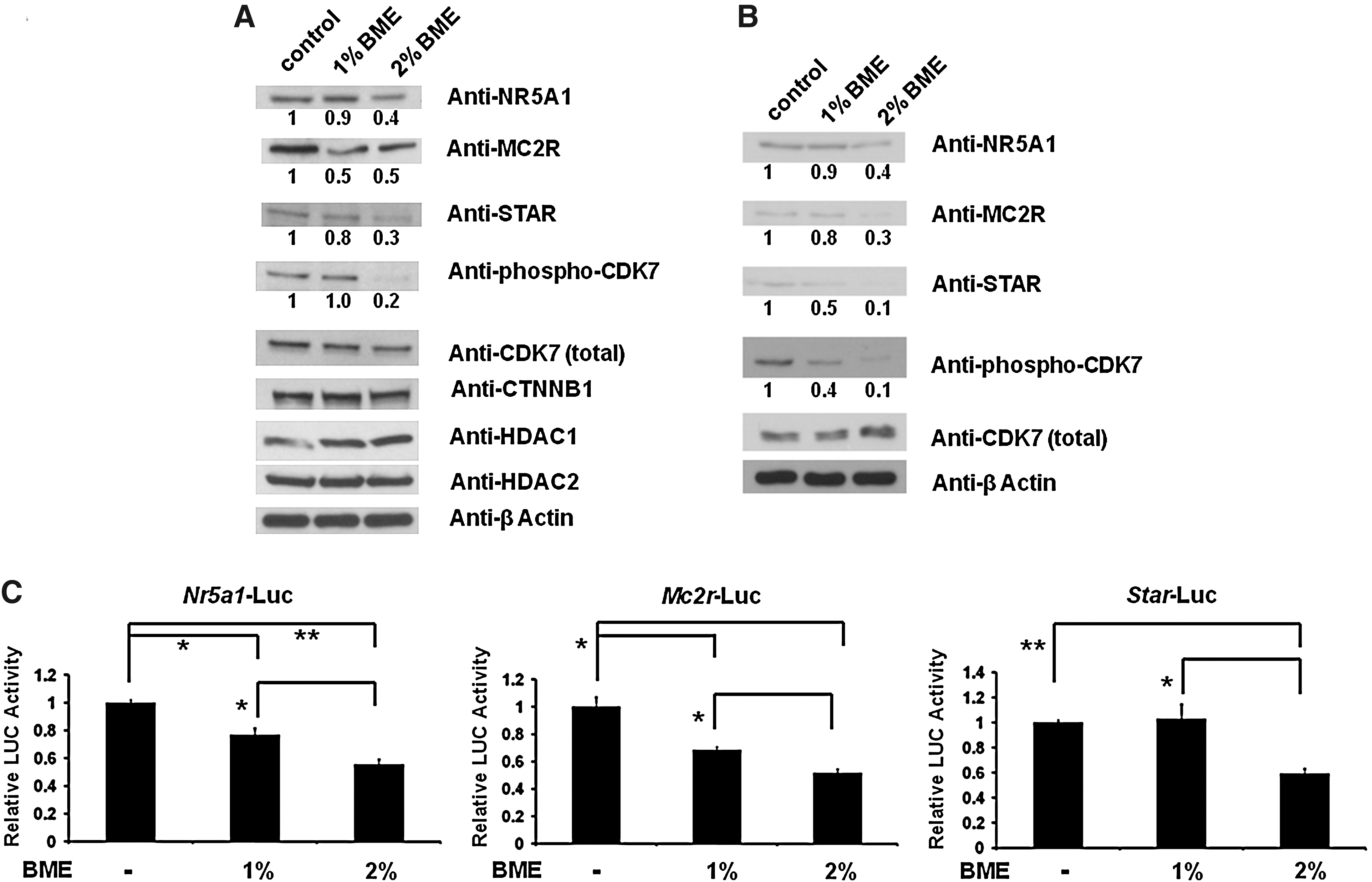
BME inhibits key regulatory proteins involved in steroidogenesis.
BME inhibits IGF1R and its downstream signaling
Recent studies have shown that overexpression of IGF2 and IGF1R plays a critical role in adrenocortical cancer pathophysiology. 4,5 Thus anti-IGF1R inhibitors and/or antagonists provide a promising therapy for treating human adrenocortical cancer. Therefore, we next evaluated the effect of BME on IGF1R level and its downstream signaling pathways in adrenocortical cancer cells. As shown in Figure 5A and B, BME treatment in both Y1 and H295R cells reduced IGF1R protein levels. Because of the importance of the phosphoinositol 3-kinase/AKT and ERK signaling pathways to the activation of IGF1R, we determined the effect of BME on the activation of these pathways. Results (Fig. 5A and B) revealed that treatment with BME at 1% (vol/vol) and 2% (vol/vol) significantly reduced AKT activation. However, the phosphorylation of ERK was slightly activated after BME treatment. In addition to reduction in IGF1R level by BME, we further observed that the level of IGF2 in Y1 cells was reduced by BME (reporter gene assay) (Fig. 5C). These results suggest that BME inhibits IGF2 synthesis and IGF1R protein level and selectively targets the AKT pathway in adrenocortical cancer cells. When these data are taken together, they suggest BME could be used to block the IGF1R/AKT pathway to prevent growth of adrenocortical cancers.

BME inhibits insulin-like growth factor 1 receptor (IGF1R)/RAC-α serine/threonine-protein kinase (AKT) signaling pathway in adrenocortical cancer cells.
Discussion
Adrenocortical cancer is a form of aggressive disease, and the prognosis rate is extremely poor. 26,27 Cancer cells usually outgrow normal cells by activating cell cycle machinery and reducing cell death. 28 Therefore, the approaches to reduce cell cycle progression and activate apoptosis to cancer cells are beneficial for cancer treatment. For centuries, humans have used natural sources such as fruits and vegetables from plants to combat and treat different forms of diseases such as cancers and diabetes. Bitter melon has received much attention for this reason. Herein, we show for the first time an antiproliferative effect of BME in adrenocortical cancer cells. BME exerts a significant effect on inhibition of cell growth and cell cycle regulation, reduction of steroidogenesis, and suppression of the IGF1R/AKT signaling pathway in adrenocortical cancer cell lines.
The anticancer effects of bitter melon have been extensively studied in recent years. First, BMEs in drinking water inhibit the development of mammary tumors in mice. 17 Second, eleostearic acid from bitter melon seeds blocks breast cancer proliferation and induces apoptosis through an oxidation-dependent mechanism. 18 Third, BME induces cell cycle arrest and promotes apoptosis in MCF-7 and MDA-MB-231 breast cancer cells. 19 Moreover, leaf extracts from bitter melon have been shown to suppress rat prostate cancer progression. 16 Several lines of evidence also suggest that eleostearic acid from bitter melon or crude BME triggers apoptosis in colon cancer cells. 20 –22 Overall, these results suggest that bitter melon has the potential to prevent the development of many different types of cancers and/or to reduce cancer progression. Therefore, the finding in our current study that BME inhibits proliferation of adrenocortical cancer cells by several different transduction mechanisms and pathways adds a new layer of information to the antitumor activities of bitter melon.
We have examined the effect of BME in two adrenocortical cancer cell lines (H295R and Y1) and primary rat adrenal cells. BME exhibited the highest antiproliferative effect against adrenocortical cancer cells while having a low-to-mild effect on primary rat adrenal cells. This indicates that BME is well tolerated and nontoxic to normal cells. Our result supports the previous conclusion that BME is relatively safe in acute and chronic doses in animal studies. 29 We further compared the antiproliferative effect of extracts from different fruits in adrenocortical cancer cells. Blueberry extracts have been shown to inhibit growth and induce cell death of breast cancer cells such as MDA-MB-231 cells, 24 but they do not display an antiproliferative effect in adrenocortical cancer cells. Therefore, BME was chosen for further study.
In the present study, we have shown that BME induced cell death and triggered apoptosis by activating proteins of the CASP family and PARP cleavage and by inhibiting critical cell cycle regulatory proteins from the CCND family. We have also observed up-regulation of TP53 and ATF3 levels in adrenocortical cancer cells treated with BME. Our results support the finding in a previous study 19 that BME exerts a significant inhibitory effect on cell growth of breast cancer cells (MCF-7 and MDA-MB-231) through the modulation of cell cycle regulation and apoptosis, suggesting the importance and significance of BME-mediated inhibition of cancer growth. Therefore, future studies are necessary to determine the anticancer effects of BME in adrenocortical cancer in vivo animal models.
Adrenocortical cells require adrenocorticotropic hormone stimulation for tropic growth. 30,31 The binding of adrenocorticotropic hormone to MC2R promotes the activation of protein kinase A and MAPK-dependent signaling cascades that collectively initiate the adrenal-specific, steroidogenic transcriptional network. 32,33 NR5A1 and STAR proteins play an essential and decisive role for steroid biosynthesis in adrenal glands and gonads. The results from the present study, for the first time, show that BME suppresses the activities of these key proteins (NR5A1, MC2R, and STAR and phosphorylation of CDK7) in steroidogenesis, suggesting that multiple mechanisms are involved in the inhibitory effect of cell growth by BME. Inhibition of cell proliferation by BME could be due to the impairment of adrenocorticotropic hormone signaling and suppression of steroidogenesis in addition to apoptosis and cell cycle alteration. It is interesting that we did not observe changes in HDAC1, HDAC2, and CTNNB1 levels in adrenocortical cancer cells treated with BME. However, the previous report showed that ribosome-inactivating proteins isolated from bitter melon inhibited HDAC1 activity in prostate cancer cells. 14 The different observation could be, at least in part, due to different cell lines used in those studies.
Most of the cancers rely on several mitogenic and angiogenic factors for rapid growth and invasion. 34 –36 In adrenocortical cancers, several molecular modifications are frequently observed, such as overexpression of vascular endothelial growth factor 37,38 and/or IGF2 4,5,26,39 and somatic mutations of TP53. 26,40,41 Recently, two studies have simultaneously shown that IGF2 and IGF1R are abundantly expressed in pediatric and adult adrenocortical carcinoma, suggesting the important role of IGF1R signaling in the development and growth of adrenocortical cancers. 4,5 Therefore, several targeted therapies, such as IGF1R antibodies or inhibitors, tyrosine kinase inhibitors, and other anti-angiogenic compounds, are now under intensive investigation and may lead to new prevention and treatment options. 38,42 In the current study, we have demonstrated that IGF2 synthesis, IGF1R level, and its downstream signaling (phosphorylation of AKT) were reduced in adrenocortical cancer cells with BME treatment. Moreover, we observed mammalian target of rapamycin transcriptional activity (by reporter gene assay) was also decreased in adrenocortical cancer cells treated with BME (data not shown). Therefore, inhibition of the IGF1R/AKT/mammalian target of rapamycin pathway by BME could be considered as a novel prevention strategy for the malignant adrenal tumors. Based on our results, we thus propose a summary showing BME-mediated antitumor effect in adrenocortical cancer cells as diagrammed in Figure 6.
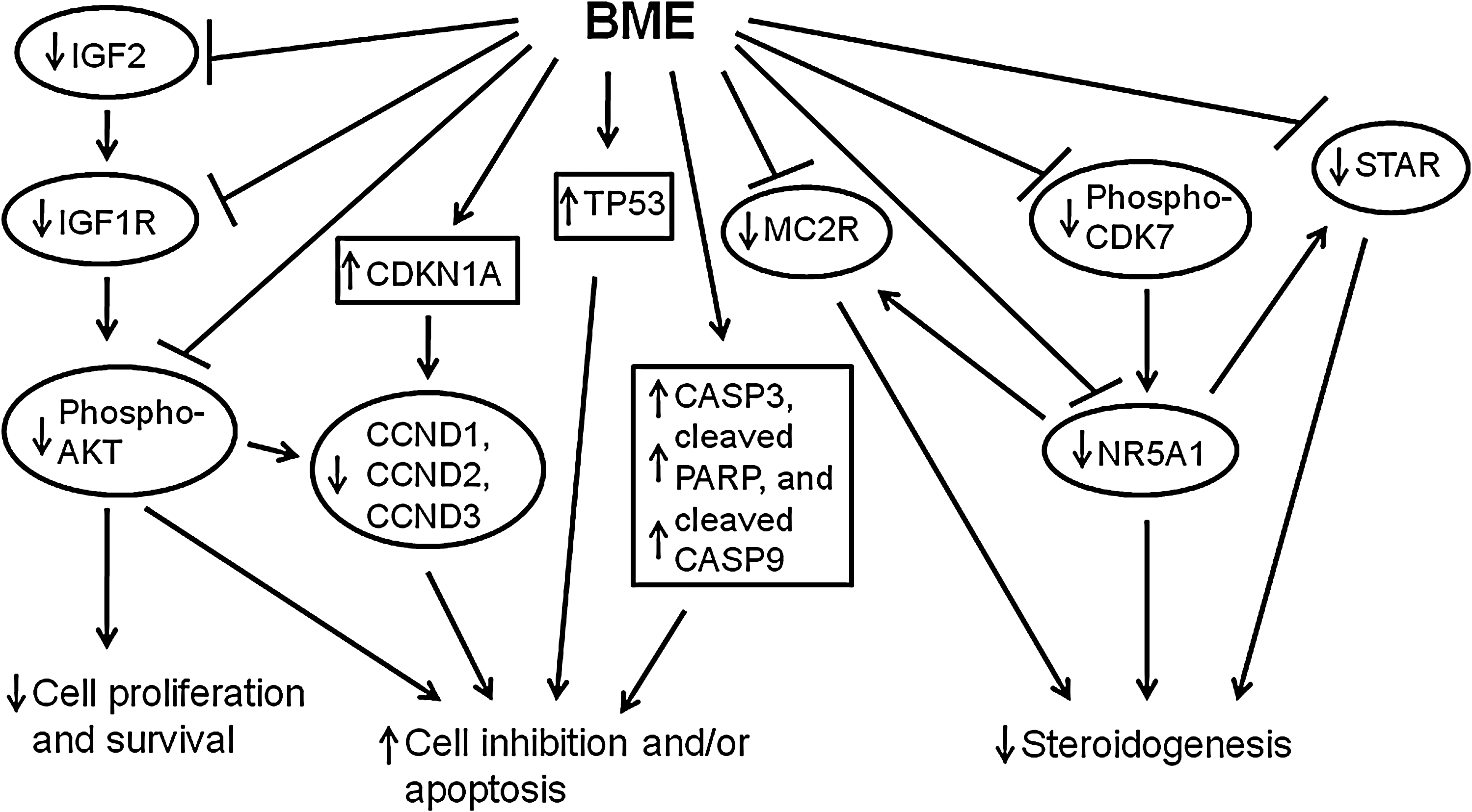
Proposed summary demonstrating BME-mediated inhibition of cell growth, alteration of cell cycle regulation, suppression of steroidogenesis, and inhibition of IGF1R/AKT signaling in adrenocortical cancer cells.
A limitation of this study is that we used crude BME instead of isolated compounds from BME in cell culture system. We chose crude BME for this study mainly because strong evidence from blueberry studies has suggested that whole extract exhibited higher antiproliferative activity than fractions or compounds. 24 The isolated compounds are usually prone to degradation and oxidation when purified. BME contains a variety of proteins, lipids, and inorganic compounds, such as ribosome-inactivating proteins, charantin, vicine, polypeptide-p, eleostearic acid, stearic acid, momordin, and Kuguacin J. 14 –16,18,43 Therefore, future studies are necessary to determine which compound or compounds of BME are involved in antiproliferation of adrenocortical cancers. The results of our current studies suggest at least in part that the oral intake of bitter melon could be used as a key component for adrenocortical cancer prevention strategies.
In this report, we have demonstrated the inhibitory effect of bitter melon on the proliferation of adrenocortical cancer cells through modulation of the apoptosis, cell cycle regulation, steroidogenesis, and IGF1R/AKT signaling. These data raise the prospect of using reduction of IGF1R/AKT signaling and induction of cell death by BME to attain a preventive advantage for adrenocortical cancers. In summary, we have identified a novel antitumor effect of BME in adrenocortical cancers. Our study adds a new layer to the previous understanding of how bitter melon functions as a folk medicine option to prevent and/or treat a variety of diseases.
Footnotes
Acknowledgments
The authors thank Dr. Kenneth Escudero (Texas A&M University, Kingsville, TX, USA), Dr. Junga Kawauchi, Drs. Costa and Raychandhuri (University of Illinois, Chicago, IL, USA), and Dr. Murrell (Cambridge Research Institute, Cambridge, United Kingdom) for providing the plasmids. The authors thank Dr. Zhi-Qing Zhao (Mercer University School of Medicine, Savannah, GA, USA) for providing normal rat adrenal glands and Drs. Buffy Ellsworth (Southern Illinois University, Carbondale, IL, USA) and Lizhong Wang (University of Michigan, Ann Arbor, MI, USA) for critical input and statistical analysis, respectively, of the manuscript. This work was supported by a Mercer University Seed Grant (to W.-H.Y.).
Author Disclosure Statement
The authors have nothing to disclose.