
Abstract
Rehmanniae Radix Preparata, the steamed root of Rehmannia glutinosa Libosch, has been widely used for the treatment of inflammatory conditions in Oriental medicines. In this study we evaluated the effects of 2,5-dihydroxyacetophenone (DHAP) isolated from Rehmanniae Radix Preparata on inflammatory responses in lipopolysaccharide (LPS)-stimulated RAW264.7 mouse macrophages. LPS-stimulated RAW264.7 cells were used to investigate the anti-inflammatory activity of DHAP on the production of inflammatory mediators such as nitric oxide (NO), inducible NO synthase (iNOS), tumor necrosis factor-α (TNF-α), and interleukin (IL)-6. DHAP significantly inhibited NO production via the suppression of iNOS expression and significantly decreased levels of the pro-inflammatory cytokines TNF-α and IL-6 via the down-regulation of their mRNA expression in LPS-stimulated RAW264.7 cells. DHAP potently inhibited the phosphorylation of extracellular signal-related kinase (ERK) 1/2 and the nuclear translocation of nuclear factor-κB (NF-κB) p65 in LPS-stimulated cells. These results indicate that DHAP inhibits the production of inflammatory mediators in activated macrophages by blocking the ERK1/2 and NF-κB signaling pathways. Our results suggest that DHAP from Rehmanniae Radix Preparata has anti-inflammatory activity in activated macrophages, raising the possibility that this compound has a therapeutic potential for inflammatory conditions.
Introduction
I
Recently, many investigations have evaluated herbal medicines and their anti-inflammatory activities at the cellular or molecular level to develop novel therapeutic strategies to suppress the production of pro-inflammatory cytokines.
3,9
The steamed root of Rehmannia glutinosa Libosch (Rehmanniae Radix), a traditional Chinese medicine, has been used to treat inflammatory conditions in Oriental clinics. This herb is known to have biological activities such as anti-inflammation
10
and wound healing
11
effects and to ameliorate diabetic nephropathy.
12
We reported the isolation of the several kinds of compounds such as 2,5-dihydroxyacetophenone (DHAP), 5-hydroxymethyl furonic acid, pomonic acid, jio-cerebrisode, and 5-(α-
In this study, we evaluated the anti-inflammatory activities of DHAP isolated from Rehmanniae Radix on the production of inflammatory mediators including NO and the pro-inflammatory cytokines TNF-α and IL-6 in lipopolysaccharide (LPS)-stimulated RAW264.7 mouse macrophages. We also identified its action mechanism via the regulation of the ERK1/2 and NF-κB pathways.
Materials and Methods
Plant material
The roots of R. glutinosa were cultivated at Buk-who-myeon, Andong-si, Gyeongbuk Province, Korea, harvested in 2008, and authenticated by Dr. J.H. Lee, a medicinal botanist in the Department of Herbology, College of Oriental Medicine, Dongguk University. The classical processing method used to prepare steamed R. glutinosa is “steaming nine times and drying in the sun nine times.” 14 The preparation was produced by steaming the roots in the crude drug manufacturing company Dae Kyung, with the procedure according to the Pharmacopoeia of Korea. A voucher specimen (10C1001-A91BXX0812) was deposited in the Korea Food & Drug Administration, Seoul, Korea.
Isolation of DHAP
The steamed root of R. glutinosa (10 kg) was extracted three times in 70% ethanol at room temperature for 7 days. After the ethanol extract was concentrated by evaporation, this extract was suspended in distilled water and partitioned with the same volume of ethyl acetate. The ethyl acetate fraction was successively partitioned with n-hexane, methanol, and water at ratio of 10:9:1 to yield n-hexane and 90% methanol fractions. The 90% methanol fraction was chromatographed on a silica gel column and eluted with a gradient of n-hexane and ethyl acetate, with subsequent repeated chromatography of this fraction over a silica gel column with 100% methylene chloride. The fraction obtained was chromatographed on a silica gel column eluted with a gradient of methylene chloride, methanol, and distilled water at a ratio of 7:0.1:0.1 to give DHAP (5 mg) as yellowish needles (yield of 60 mg), which was identified by spectroscopic data: ultraviolet, λmax (log ɛ) (methanol)=226 (4.33), 256 (3.97), 363 (3.75) nm;
1
H-nuclear magnetic resonance (400 MHz, CD3OD) δ, 6.78 (1H, d, J=8.9 Hz, H-3), 6.99 (1H, dd, J=2.9, 8.9 Hz, H-4), 7.21 (1H, d, J=2.9 Hz, H-6);
13
C-nuclear magnetic resonance (100 MHz, CD3OD) δ, 121.6 (C-1), 157.3 (C-2), 120.4 (C-3), 126.7 (C-4), 151.4 (C-5), 117.2 (C-6), 206.7 (

Chemical structure of 2,5-dihydroxyacetophenone.
Cell culture
RAW264.7 mouse macrophages (American Type Culture Collection, Rockville, MD, USA) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT, USA) in a 5% CO2 humidified atmosphere at 37°C. Prior to each experiment, cells were plated in 24-well plates at a cell density of 1×105 /mL for culture supernatant tests or in 60-mm-diameter dishes at a cell density of 1×106 /mL for protein extraction and RNA extraction. The next day, the cells were pretreated with various concentrations of DHAP for 1 h and then stimulated with 1 μg/mL LPS (serotype O111:B4, 10 mg/mL; Sigma Aldrich, St. Louis, MO, USA) for the indicated time interval.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay for cell viability
Cell viability was assessed by the conversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Roche, Mannheim, Germany) in the MTT reduction assay. The cells were pretreated with different concentrations of DHAP (10, 50, 100, and 200 μM) for 30 min prior to stimulation with LPS (1 μg/mL) for 24 h. At the termination of cultures, 10 μL of MTT solution was added to each well, and the cells were then cultured for 4 h. One hundred microliters of dimethyl sulfoxide was added to each well, and then the optical density was measured at 550 nm by a microplate reader (GENios, TECAN Instruments, Inc., Grödig, Austria).
Determination of NO production
The cells (1×105 /mL) were pretreated with different concentrations of DHAP for 30 min and then stimulated with LPS (1 μg/mL) for 24 h. The culture media were collected and assayed for NO production using Griess reagents [1% sulfanilamide and 0.1% N-(1-naphthyl)-ethylenediamine dihydrocholoride in 5% phosphoric acid] (Roche). The absorbance at 540 nm was determined using a microplate reader. A standard curve was generated in the same fashion using NaNO2.
Measurement of cytokine levels
The concentrations of TNF-α and IL-6 in each culture medium were measured by commercially available enzyme-linked immunosorbent assay kits according to the manufacturer's instruction (eBioscience, San Diego, CA, USA). The concentration of each cytokine was calculated according to the equation obtained from a standard curve using the standard solution in the enzyme-linked immunosorbent assay kit.
RNA extraction and reverse transcription–polymerase chain reaction
The cells were pretreated with different concentrations of DHAP for 30 min prior to stimulation with LPS (1 μg/mL) for 4 h. Total RNA from each cell was isolated using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer's instructions. Total RNA was reverse-transcribed for 1 h at 42°C in a reaction mixture containing RNA, 1× reverse transcriptase buffer (Promega, Madison, WI, USA), 0.5 mM deoxynucleotide triphosphates, 3 mM MgCl2, 5 U of RNase inhibitor (Amersham Bioscience, Piscataway, NJ, USA), 0.5 μM oligo(dT) primer, and 5 U of SuperScript® reverse transcriptase (Promega) in a total volume of 20 μL. The polymerase chain reaction (PCR) was performed using the above prepared cDNA as a template with the following cycle parameters: 94°C, 2 min, 30–35 cycles; 94°C, 30 s; 56–59°C, 30 s; 72°C, 1 min; and 92°C, 10 min. PCR products were then electrophoresed on 1% agarose gels at 100 V. Specific genes were verified by assessing their predicted sizes under ultraviolet light. The primer sequences were as follows: cyclooxygenase-2 (accession number 011198), forward 5′-CAC TAC ATC CTG ACC CAC TT-3′, reverse 5′-ATG CTC CTG CTT GAG TAT GT-3′; TNF-α (accession number 013693), forward 5′-CTC AGA TCA TCT TCT CAA AAT TCG AGT GAC A-3′, reverse 5′-CTT CAC AGA GCA ATG ACT CCA AAG T-3′; IL-1β (accession number 008301), forward 5′-ATG GCA ACT GTT CCT GAA CTC AAC T-3′, reverse 5′-CAG GAC AGG TAT AGA TTC TTT CCT TT-3′; IL-6 (accession number NM03116.8), forward 5′-TTC ACA GAG GAT ACC ACT CC-3′, reverse 5′-TAG CCA CTC CTT CTG TGA CT-3′; iNOS (accession no. 010927), forward 5′-TTT GGA GCA GAA GTG CAA AGT CTC-3′, reverse 5′-GAT CAG GAG GGA TTT CAA AGA CCT-3′; and glyceraldehyde 3-phosphate dehydrogenase (accession number XM 994067.2), forward 5′-CTC CTG GAG TCT ACT GGT GT-3′, reverse 5′-GTC ATC ATA CTT GGC AGG TT-3′. Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control for PCR.
Western blot analysis
The cells were pretreated with different concentrations of DHAP for 30 min prior to stimulation with or without 1 μg/mL LPS for 24 h. Cells were lysed with lysis buffer (10 mM Tris-HCl [pH 7.9], 10 mM NaCl, 3 mM MgCl2, and 1% Nonidet P-40). For nuclear extraction, cells were washed twice with cold phosphate-buffered saline and lysed with NE-PER™ nuclear and cytoplasmic extraction reagents (Pierce, Rockford, IL, USA). After centrifugation at 14,000 g for 10 min, the supernatant was stored at −80°C until use. The protein concentration was determined by Bradford's assay. Protein (30 μg/mL) was separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. The membranes were blocked with 5% skim milk (BD, Franklin Lakes, NJ, USA) in Tris-buffered saline with Tween (10 mM Tris-HCl, 150 mM NaCl, and 0.5% Tween-20) for 1 h. The membranes were incubated overnight with primary antibodies at 4°C and then incubated with horseradish peroxidase–conjugated secondary antibodies. The blots were developed with ECL western detection reagents (Amersham Bioscience). The antibodies used in this study were anti-iNOS (diluted 1:1000, Cell Signaling Technology, Beverly, MA, USA), anti-β-actin (diluted 1:1000, Sigma Aldrich), anti-TATA binding protein (diluted 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phospho-ERK 1/2 (diluted 1:1000, Cell Signaling), anti-phospho-p38 (diluted 1:1000, Cell Signaling), anti-phospho-stress-activated protein kinase/c-Jun N-terminal kinase (JNK) (diluted 1:1000, Cell Signaling), anti-NF-κB (diluted 1:1000, Cell Signaling Technology), and horseradish peroxidase–labeled anti-rabbit or mouse immunoglobulin G (diluted 1:5000; Santa Cruz Biotechnology).
Statistical analysis
The results of all experiments were expressed as mean±SD values and are representative of three independent experiments. Statistical analysis was carried out by one-way analysis of variance with the post hoc test using PRISM version 5.0 statistical analysis software (GraphPad Software, Inc., San Diego). Values of P<.05 were considered significant.
Results
Effect of DHAP on cell viability
To avoid any cytotoxic effects caused by DHAP, we investigated the effects of DHAP on cell viability in RAW264.7 cells by MTT assay (Fig. 2). The results showed that treatment with DHAP at concentrations of 10, 50, and 100 μM did not cause cell death. Therefore, we used DHAP at concentrations of 100 μM or less for further studies of its anti-inflammatory properties and action mechanism in the cells.

Effect of 2,5-dihydroxyacetophenone (DHAP) on cell viability in RAW264.7 cells. Cells were incubated with DHAP for 24 h, and then cytotoxicity was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Data are mean±SD values (n=3). *P<.05 versus normal (0 μM DHAP).
Effect of DHAP on LPS-induced NO production
Because NO is the major inflammatory mediator synthesized by iNOS during inflammation, 15 we examined the effect of DHAP on NO production in LPS-stimulated RAW264.7 cells. As shown in Figure 3, DHAP significantly (P<.05) decreased LPS-induced production of NO in a dose-dependent manner. At the highest concentration of DHAP tested (100 μM), NO production was reduced to approximately basal levels or to that of unstimulated cells. DHAP alone did not induce NO production. Next, by reverse transcription–PCR, we investigated the effect of DHAP on the expression of iNOS mRNA in LPS-stimulated cells (Fig. 4). DHAP attenuated LPS-induced expression of iNOS mRNA, suggesting that this compound inhibits NO production at the transcriptional level of its gene in LPS-stimulated macrophage cells.
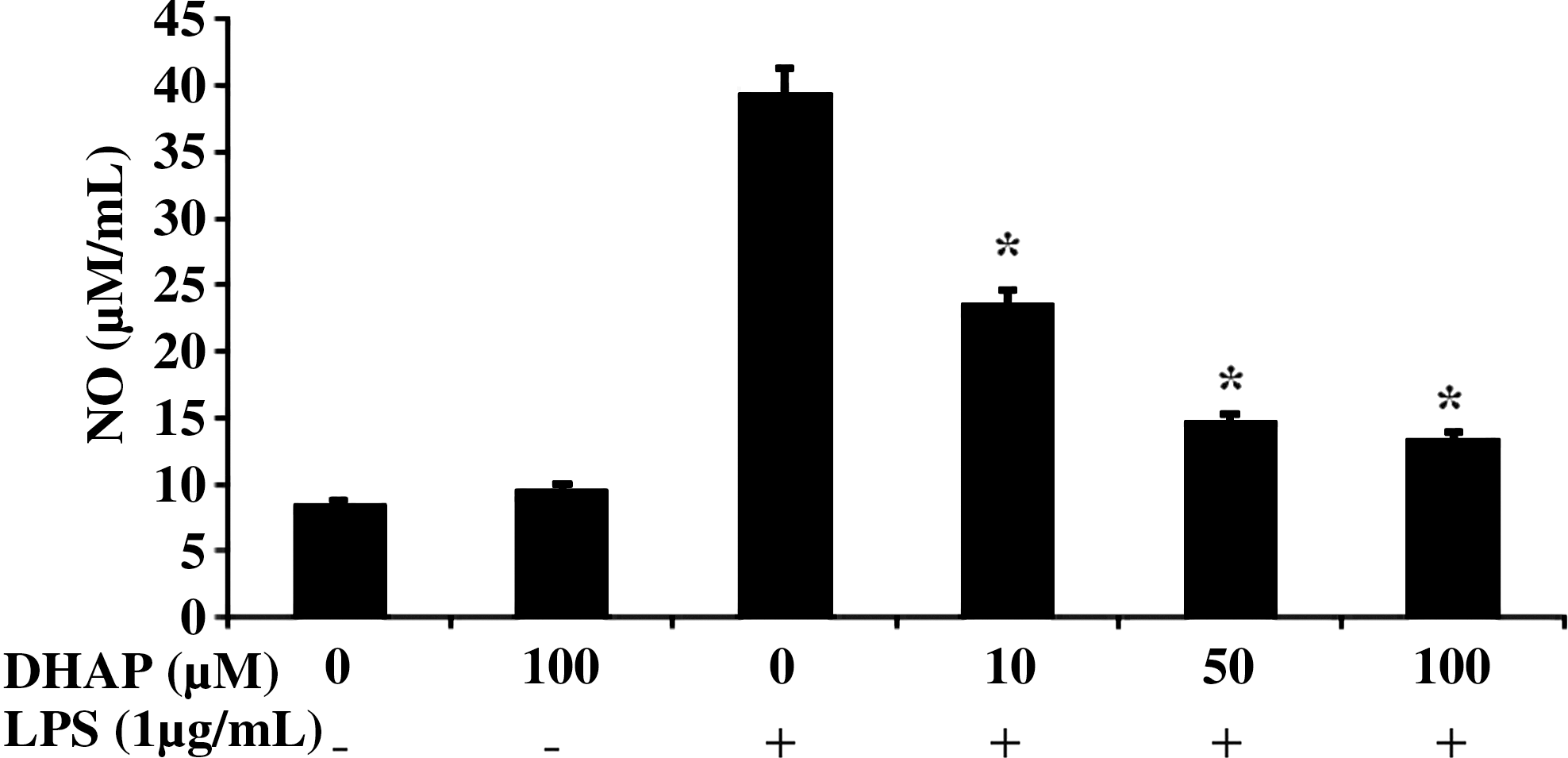
Effect of DHAP on lipopolysaccharide (LPS)-induced nitric oxide (NO) production in RAW264.7 cells. Cells were incubated with various concentrations of DHAP without or with LPS (1 μg/mL) for 24 h. NO was quantified in culture medium by the Griess reaction. Data are mean±SD values (n=3). *P<.05 versus LPS alone.
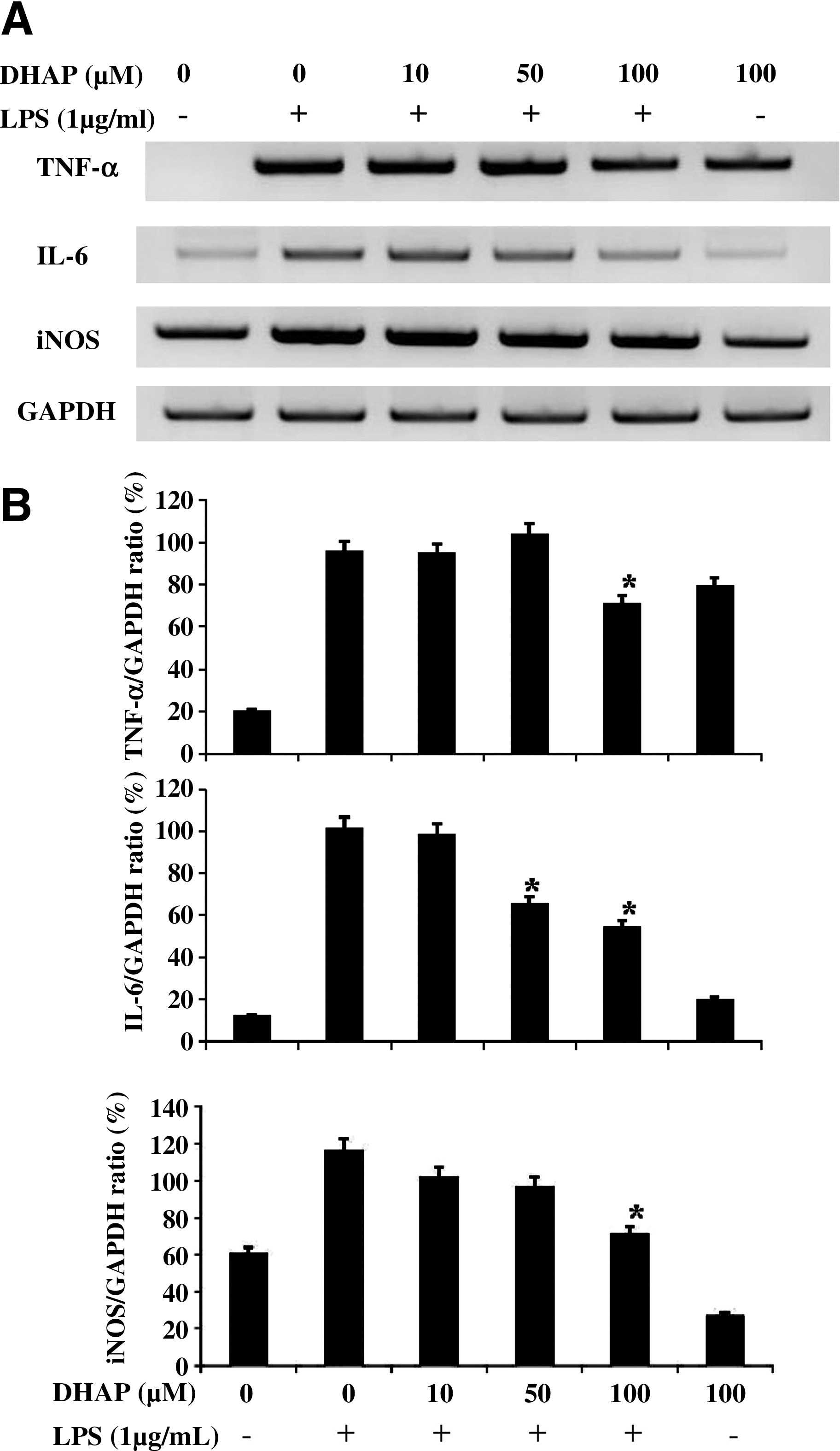
Effect of DHAP on LPS-induced expression of tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and inducible NO synthase (iNOS) mRNA in RAW264.7 cells. Cells were incubated with DHAP without or with LPS (1 μg/mL) for 4 h.
Effect of DHAP on LPS-induced production of pro-inflammatory cytokines
To determine the effect of DHAP on production of pro-inflammatory cytokines, we measured the levels of TNF-α and IL-6 in LPS-stimulated RAW264.7 cells by enzyme-linked immunosorbent assay (Fig. 5). The results showed that DHAP significantly and dose-dependently decreased LPS-induced production of TNF-α and IL-6. We also investigated whether DHAP regulated the transcription of cytokine mRNA by reverse transcription–PCR (Fig. 4) in the cells. The mRNA expression of TNF-α and IL-6 was suppressed by the treatment with DHAP. Therefore, the density ratio of TNF-α and IL-6 was significantly (P<.05) suppressed by DHAP in LPS-stimulated cells. These data suggest that DHAP prevents pro-inflammatory cytokine production by suppressing their gene expression in LPS-stimulated macrophages.
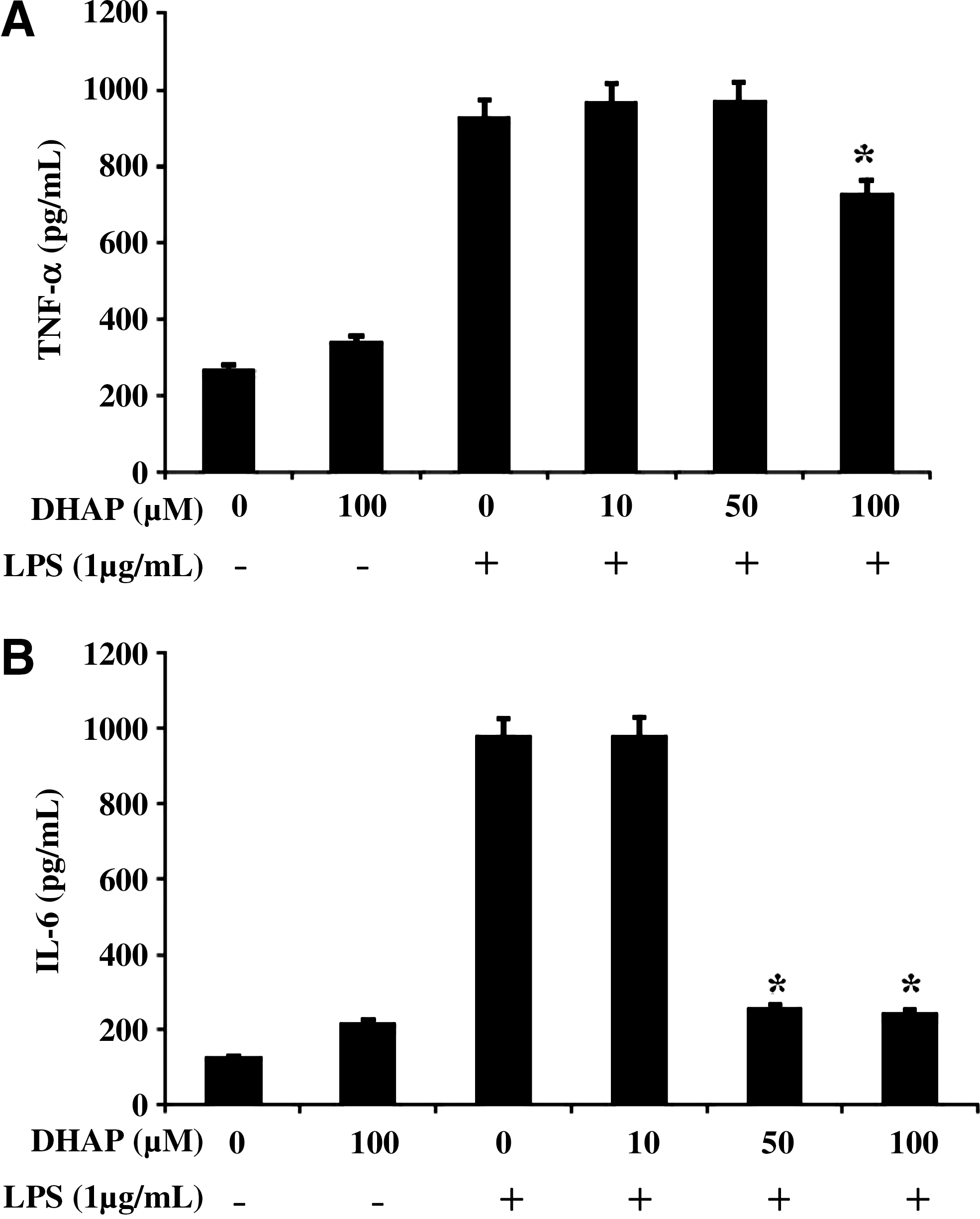
Effect of DHAP on LPS-induced production of pro-inflammatory cytokines in RAW264.7 cells. Cells were incubated with DHAP without or with LPS (1 μg/mL) for 24 h.
Effect of DHAP on LPS-induced phosphorylation of ERK1/2 and translocation of NF-κB p65
To investigate whether DHAP modulates the inflammation-mediated signal pathway in activated macrophages, we investigated the phosphorylation of ERK1/2 and the activation of NF-κB in LPS-stimulated RAW264.7 cells by western blot (Fig. 6). LPS stimulation rapidly induced ERK1/2 phosphorylation in the cells (Fig. 6A and B). However, DHAP suppressed LPS-induced phosphorylation of ERK1/2, whereas expression of the nonphosphorylated form remained the same. We next examined the influence of DHAP on NF-κB levels in the nuclei of the cells by western blot analysis (Fig. 6C and D). LPS stimulation induced the nuclear translocation of NF-κB in the cells. However, DHAP blocked LPS-induced translocation of NF-κB into the nuclei. In quantitative analysis, we also found that 50 and 100 μM DHAP had significant inhibitory effects on LPS-induced phosphorylation of cytosolic NF-κB p65. These results indicate that the signaling pathway of ERK1/2 and NF-κB is effectively blocked by DHAP in activated macrophages.
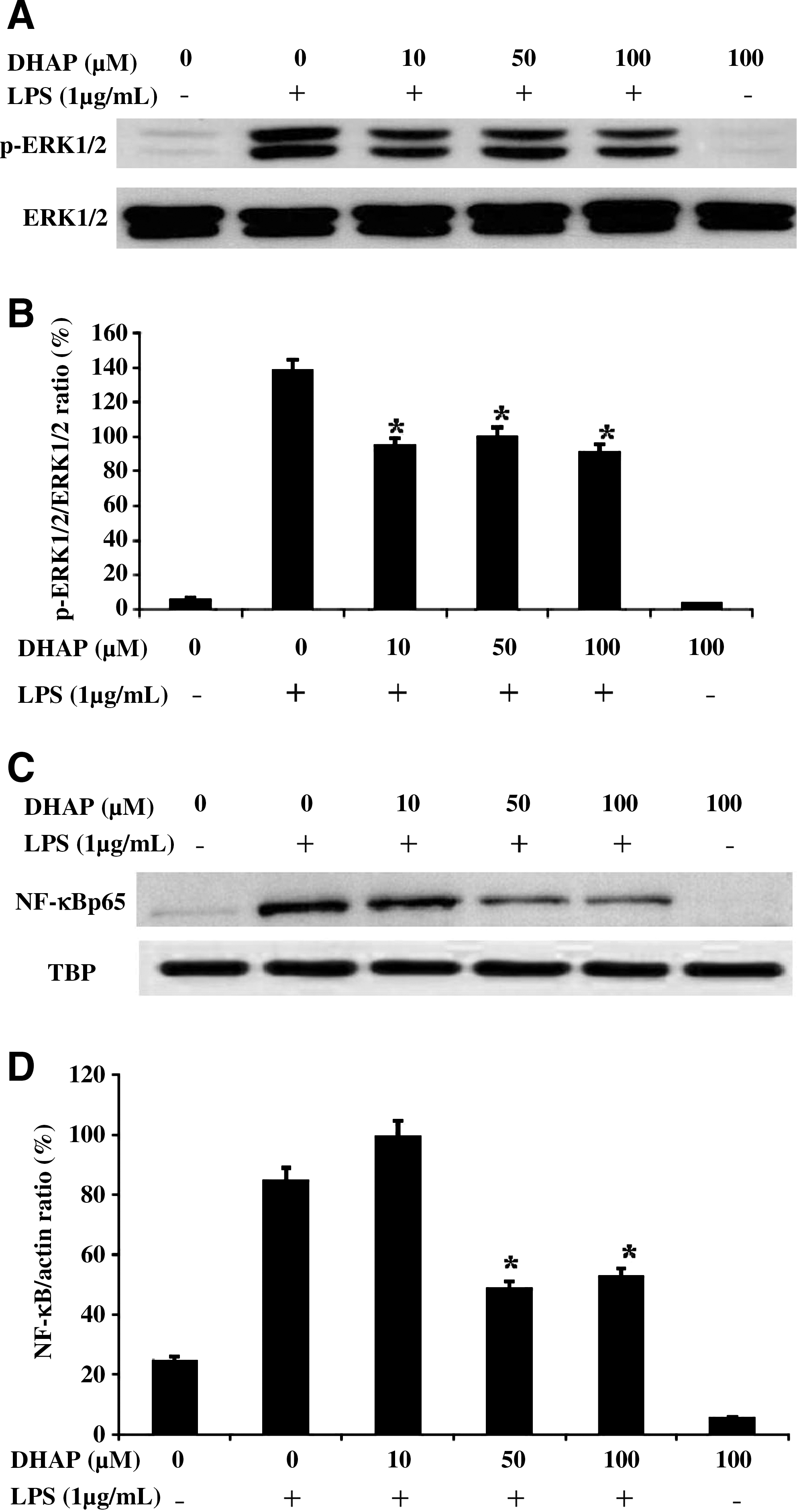
Effect of DHAP on LPS-induced phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 and nuclear factor-κB (NF-κB) translocation in RAW264.7 cells. Cells were incubated with DHAP without or with LPS (1 μg/mL) for 24 h. The
Discussion
This study was conducted to elucidate the effects of DHAP, a natural compound isolated from Rehmanniae Radix Preparata, on the production of inflammatory mediators in activated macrophages and to identify its mechanism of action. We found that DHAP effectively suppressed the production of NO, TNF-α, and IL-6 by suppressing the expression of iNOS and cytokine mRNA in LPS-stimulated RAW264.7 cells. Furthermore, the molecular mechanism by which DHAP inhibited the expression of these mediators appeared to involve the inhibition of ERK1/2 and NF-κB activation. Our results suggest that DHAP may have good anti-inflammatory activity in activated macrophages by suppressing the overproduction of inflammatory mediators.
In response to LPS, iNOS induction in macrophages leads to substantial increases in NO production, and iNOS is an important effector molecule in immune regulation and defense. 2,5 High levels of NO produced by iNOS have been defined as cytotoxic in inflammation. 16 Thus, potential inhibitors of iNOS are considered useful for the treatment of a variety of inflammatory diseases. 9 In the present study, DHAP significantly inhibited excessive NO production in LPS-stimulated macrophages. Moreover, this suppression of LPS-induced NO production was accompanied by down-regulation of iNOS transcription. LPS also induced macrophages to produce pro-inflammatory cytokines, such as TNF-α and IL-6, which are relevant in various types of inflammation; reduction in levels of these cytokines may retard inflammatory responses. 17,18 Therefore, inhibition of cytokine production or function serves as a key mechanism in the control of inflammation. 16,19 This study showed that DHAP remarkably suppressed the production and mRNA of TNF-α and IL-6 in LPS-stimulated cells. These findings provide evidence that DHAP possesses potential anti-inflammatory activity by modulating inflammatory mediator synthesis.
In addition, it is well known that LPS induces inflammatory responses such as the mitogen-activated protein kinase (MAPK) pathways that include ERK1/2, JNK, and p38 MAPKs and the NF-κB pathway. 9 The MAPKs are thought to be the most important signaling molecules that control NF-κB activation and synthesis of pro-inflammatory mediators. 20 Meanwhile, NF-κB is a key molecule in the inflammatory response in macrophages. Under normal conditions, NF-κB cannot translocate into the nucleus because of inhibition by the inhibitor of κB. However, upon LPS stimulation, the inhibitor of κB is phosphorylated and then targeted for ubiquitination and degradation, after which the activated NF-κB is able to translocate into the nucleus. 2 Thus, in this study, to investigate the modulation of upstream signaling molecules related to the NF-κB pathway, the phosphorylation of ERK1/2, JNK, and p38 MAPKs was assessed in LPS-stimulated RAW264.7 cells. Our results showed that DHAP selectively reduced the phosphorylation of ERK1/2 and blocked the nuclear translocation of NF-κB in LPS-stimulated cells. These findings suggest that DHAP possesses potential anti-inflammatory activity via regulation of NF-κB pathway-mediated inflammatory responses in activated macrophages.
In conclusion, we demonstrate that DHAP, a natural compound isolated from Rehmanniae Radix Preparata, is a potent inhibitor of the secretion of an inflammatory mediator, NO, and the pro-inflammatory cytokines TNF-α and IL-6 in LPS-stimulated macrophages. Thus, DHAP may have therapeutic potential for improvement of inflammatory conditions via the regulation of macrophage activation and may be an effective treatment option for a variety of inflammatory diseases.
Footnotes
Acknowledgment
This study was supported by the research fund of Studies on the Identification of the Efficacy of Biologically Active Components from Oriental Herbal Medicines from the Korean Food and Drug Administration (2011).
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.