
Abstract
High levels of reactive oxygen species inflict oxidative damage on various cellular components that eventually culminate in a variety of diseases. This study investigated the cytoprotective effects of a nucleic acid–based health product (Squina [Hong Kong, China] DNA) against oxidative stress, particularly in neuronal cells. Adult female Sprague–Dawley rats were treated with Squina DNA, and changes in mitochondrial antioxidant status and functional capacity were assessed by the activities of antioxidant enzymes and ATP generation capacity in brain, heart ventricular, kidney, skeletal muscle, and liver tissues of control and Squina DNA–treated rats. The effects of Squina DNA pretreatment on brain/neuronal cell injury were investigated in a rat model of cerebral ischemia/reperfusion (I/R) injury and a neuroblastoma SH-SY5Y cell model of β-amyloid (Aβ) protein fragment 25–35-induced toxicity. Long-term Squina DNA treatment caused dose-dependent increases in mitochondrial antioxidant status and functional capacity in rat brain, heart ventricular, kidney, skeletal muscle, and liver tissues. Squina DNA pretreatment significantly prevented I/R injury in brain tissue. The cerebroprotection was associated with a reversal of I/R-induced impairment in mitochondrial antioxidant status and disruption in membrane integrity. Squina DNA ethanol extract also significantly inhibited the Aβ-induced apoptosis in SH-SY5Y neuronal cells, as evidenced by less caspase 3 and caspase 9 activation as well as mitochondrial cytochrome c release in Aβ-challenged cells. Squina DNA may enhance the resistance of tissues and cells to oxidative stress, particularly in pathological conditions such as stroke and aging-related neurodegenerative diseases.
Introduction
R
Recent experimental and clinical studies have demonstrated that the consumption of salmon milt enhances hepatic antioxidant status by increasing superoxide dismutase and catalase activities or enhancing the oxidation–reduction cycle of coenzyme NAD of the antioxidant machinery in mice. 3,4 Squina (Hong Kong, China) DNA, a nucleic acid–based health product derived from salmon milt, is also claimed to protect DNA from oxidation. 5 In the present study, we investigated whether Squina DNA can improve mitochondrial antioxidant status and ATP generation capacity (ATP-GC) in various tissues (including brain, heart, kidney, liver, and skeletal muscle) of rats. Among all the organs tested, the brain is most susceptible to ROS-mediated damage. On the one hand, brain tissue is highly enriched in nonheme iron that catalytically participates in the production of ROS, 6 and the high level of polyunsaturated fatty acids in brain tissue renders it particularly vulnerable to ROS-induced peroxidation. 7 However, brain tissue has a relatively low level of antioxidant enzymes, such as catalase and glutathione peroxidase (GPX), to defend against oxidative challenge. 8 Therefore, it will be of pharmacological interest to investigate whether or not Squina DNA can enhance mitochondrial antioxidant status and functional capacity in brain tissue.
In the present study, an in vivo rat model of cerebral ischemia/reperfusion (I/R) injury and an in vitro neuroblastoma SH-SY5Y cell model of β-amyloid (Aβ) protein fragment 25–35-induced toxicity were utilized to investigate the effect of Squina DNA pretreatment on brain/neuronal cell injury. The development of cerebral I/R injury, which resembles ischemic stroke, involves ROS-mediated processes. 9 The accumulation of aggregated Aβ peptides in the brain of patients suffering from Alzheimer's disease causes chronic oxidative stress throughout the disease progression, with resultant mitochondrial-dependent neurodegeneration. 10 In this regard, Aβ protein fragment 25–35, which contains a functional domain of full-length Aβ required for both neurotrophic and neurotoxic effects, has been widely used as an Alzheimer's disease–inducing agent. The two experimental models adopted in the present study are representative of acute and chronic oxidative challenges to brain tissue and neuronal cells, respectively.
To elucidate the biochemical mechanism(s) underlying the cerebroprotection by Squina DNA against I/R injury, mitochondrial antioxidant status, as assessed by levels of reduced glutathione (GSH) and malondialdehyde (MDA) (a product of lipid peroxidation), 11 the sensitivity of mitochondria to Ca2+-induced permeability transition (PT), and the extent of mitochondrial cytochrome c release were measured in control and ischemic/reperfused brain tissue in rats, without and with Squina DNA pretreatment. The level/extent of cellular GSH, ROS production, activation of caspase 3 and 9, and cytochrome c release were also examined in control and Squina DNA–pretreated SH-SY5Y neuronal cells, with or without Aβ challenge.
Materials and Methods
Chemicals and Squina DNA ethanol extract
All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA) unless otherwise specified. All chemicals were of analytical grade. Squina DNA was manufactured by Maruha Corp. (Tokyo, Japan) and supplied by Squina International Group, Ltd. According to the manufacturer's specification, the product contains salmon milt–derived DNA (14%, wt/wt) and yeast-derived RNA (6%). The ethanol extract of Squina DNA (250 mg of DNA and 100 mg of RNA per 500 mg of Squina DNA) was used for experiments involving cultured SH-SY5Y cells in order to obtain a detectable response with higher tested concentrations. In brief, Squina DNA (152.8 g), which was extracted twice with 500 mL of ethanol under heat reflux for 2 h, yielded 9.9 g of Squina DNA ethanol extract. Spectrophotometric analysis indicated that the Squina DNA ethanol extract contained 3.6% (wt/wt) DNA/RNA. The ethanol extract was dissolved in dimethyl sulfoxide at a concentration of 150 mg/mL and stored at −20°C until use.
Animal and Squina DNA pretreatment
Adult female Sprague–Dawley rats (approximately 10 weeks old; weighing 250–300 g) were maintained in a 12-h dark/light cycle at about 22°C and allowed food and water ad libitum. Animals were randomly divided into three groups, with five animals in each: Group 1, vehicle control; Group 2, DNA Low Dose (18 mg/kg)×14 doses; and Group 3, DNA High Dose (54 mg/kg)×14 doses. Animals were sacrificed 24 h after the last dosing, and the whole brain, heart ventricular, kidney, skeletal muscle, and liver tissues were removed with the animal under phenobarbital anesthesia for analysis.
Preparation of tissue homogenates
Brain homogenates were obtained by homogenizing minced whole brain tissues (∼2 g) in 10 mL of ice-cold sucrose buffer (0.25 M sucrose, 0.1 mM Na2EDTA, and 5 mM Tris-HCl, pH 7.4) with a Teflon® (Dupont, Wilmington, DE, USA)–glass homogenizer at 200 rpm for 8–10 complete strokes.
Heart homogenates were obtained by homogenizing minced heart ventricular tissues (∼0.6 g) in 10-fold (wt/vol) ice-cold sucrose buffer (0.32 M sucrose, 1 mM Na2EDTA, and 50 mM Tris-HCl, pH 7.4) with a Teflon–glass homogenizer at 4000 rpm for 25–30 complete strokes.
Kidney homogenates were obtained by homogenizing minced kidney tissues (∼0.6 g) in 6 mL of ice-cold sucrose buffer (0.25 M sucrose, 0.1 mM Na2EDTA, and 5 mM Tris-HCl, pH 7.4) with a Teflon–glass homogenizer at 2000 rpm for 8–10 complete strokes.
Liver homogenates were obtained by homogenizing minced liver tissues (0.6 g) in 6 mL of ice-cold sucrose buffer (0.25 M sucrose, 0.1 mM Na2EDTA, and 5 mM Tris-HCl, pH 7.4) with a Teflon–glass homogenizer at 2000 rpm for 8–10 complete strokes.
Muscle homogenates were obtained by mixing minced skeletal muscle tissues (∼2.5 g) with 10 mL of collagenase solution (0.075% [wt/vol] in buffer), and the mixtures were incubated at 4°C for 20 min. The digested tissue mixtures were centrifuged at 600 g at 4°C for 20 min. After the supernatant was removed, the digested tissues were homogenized in 20 mL of ice-cold homogenizing buffer (100 mM KCl, 50 mM MOPS, and 10 mM EGTA, pH 7.2) with a Teflon–glass homogenizer at 9,500 rpm for 10 s, and then followed by a further homogenization with a Teflon–glass homogenizer at 4000 rpm for 25–30 complete strokes.
Preparation of mitochondrial and cytosolic fractions
Mitochondrial pellets were prepared from tissue homogenates by centrifugation at 600 g at 4°C for 20 min, followed by centrifugation of the resultant supernatant at 9200 g at 4°C for 30 min. The supernatants were collected as the cytosolic fractions of various tissues. Mitochondrial pellets were then resuspended in 1 mL of the respective homogenizing buffer used for various tissues and constituted the mitochondrial fractions.
Cerebral I/R injury
Adult female Sprague–Dawley rats were orally treated with Squina DNA at a daily dose of 18 or 54 mg/kg for 14 days. The dose of 0.18 g/kg is an equivalent of a human dose that enhanced mitochondrial antioxidant status and functional capacity in rats. For the I/R groups, 24 h after the last dosing, phenobarbital-anesthetized animals were subjected to I/R challenge by clamping both carotid arteries for 85 min and then declamped for reperfusion for 45 min. By the end of reperfusion, the whole brain was harvested for quantifying I/R-induced cerebral injury or subjected to biochemical analyses. The mortality rate for rats undergoing I/R during the experimental period was almost zero.
Quantification of I/R-induced brain injury
After the I/R experiment, the brain was removed, frozen for 10 min at −20°C, and then fitted into a mold. Six coronal brain sections (6–8 mm thick) were sliced from the front pole using a razor. The sections were incubated in phosphate-buffered saline (PBS) containing 2% (wt/vol) 2,3,5-triphenyltetrazolium chloride at 37°C for 10 min and then kept in 10% (vol/vol) buffered formalin (pH 7.0) overnight. The percentage of 2,3,5-triphenyltetrazolium chloride-stained (i.e., viable) tissue with respect to the whole piece of tissue was estimated by ImageJ software (National Institutes of Health, Bethesda, MD, USA). The extent of I/R injury was measured by noting the difference between percentage of viable tissues of non-I/R and I/R animals in the fourth brain slice from the pole.
Cell culture, Squina DNA incubation, and Aβ challenge
The human neuroblastoma clonal SH-SY5Y cell line was purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) supplied with 10% fetal bovine serum (Invitrogen). Cells used for experiments were seeded at a density of 5×105 cells per well on a six-well cell culture dish or 2.5×104 cells per well of a 96-well black multititer plate with a clear bottom when measuring the ROS generation. Aβ was dissolved in sterile deionized water and stored in at −20°C until use. To obtain the neurotoxic form of Aβ, the peptide solution was placed in an incubator at 37°C for 7 days prior to the experiments. 12 Cells were incubated with Squina DNA ethanol extract at 150 or 300 μg/mL for 24 h prior to Aβ (20 or 40 μM) challenge for 48 h. Cell lysates were then prepared and subjected to biochemical analyses.
Preparation of cell lysates
SH-SY5Y cells were harvested by trypsinization, and then the cells were resuspended in lysis buffer (20 mM Tris-HCl, 2 mM EDTA, 3 mM EGTA, 1% [vol/vol] Triton X-100, 10% [vol/vol] glycerol, and 2 mM dithiothreitol, pH 7.5) and incubated on ice for 5 min. The cells were then centrifuged at 14,000 g for 10 min at 4°C, and the resultant supernatant was subjected to biochemical and western blot analysis. For measuring cytochrome c release, the cytosolic fractions of the harvested SH-SY5Y cells were prepared as previously reported.
13
In brief, cells were washed with ice-cold PBS, subsequently incubated in permeabilization buffer (210 mM
Biochemical analyses
Mitochondrial antioxidant status was assessed by measuring GSH and oxidized glutathione (GSSG) levels as well as glutathione-related antioxidant enzyme activities. Cellular GSH and GSSG levels were measured by an enzymatic method previously described, 14 and the GSH/GSSG ratio was estimated. Glutathione reductase (GR), selenium-GPX, glutathione S-transferases (GST), and isocitrate dehydrogenase (ICDH) activities were measured by monitoring the oxidation of NADPH using spectrophotometric methods, as previously described. 15 Mitochondrial MDA levels were measured using high-performance liquid chromatography. 15 Mitochondrial functional capacity was assessed by the measurement of mitochondrial ATP-GC, as previously described. 16 Mitochondrial structural integrity was assessed by measurements of Ca2+ content, as well as the sensitivity of mitochondria to Ca2+-induced PT (expressed as Ca2+-induced mitochondrial swelling indices) and cytochrome c release, as described previously. 17 Aβ-induced cell apoptosis was measured by caspase 3 activation using a commercially available assay kit (PerkinElmer, Waltham, MA, USA). ROS generation was measured using 2′,7′-dichlorofluorescein diacetate as a probe. In brief, differentiated SHSY-5Y cells cultured on a 96-well black multitier plate with a clear bottom were washed twice with PBS and then incubated with 0.1% bovine serum albumin in PBS containing 5 μM (final concentration) 2′,7′-dichlorofluorescein diacetate (dissolved in 0.2% [vol/vol] ethanol) for 30 min at 37°C. The cells were washed twice with PBS again and then incubated with fresh medium containing 500 μM NADPH (final concentration) and Squina DNA ethanol extract (150 or 300 μg/mL). The fluorescence intensity of each well was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm with a Victor V2 Multi-label Counter (Perkin Elmer, Turku, Finland) every minute for 6 h at 37°C. The fluorescence intensity of drug-treated cells was normalized with reference to the respective time-matched vehicle (i.e., dimethyl sulfoxide) control. The extent of drug-induced ROS production was estimated by computing the area under the curve plotting normalized fluorescence intensity against time (in minutes) and expressing the result in arbitrary units.
Western blot analysis
Brain cytosolic fraction (40 μg) or SH-SY5Y cell lysate (50 μg) was mixed with Laemmli loading buffer, boiled for 5 min, and separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis at 140 V followed by electroblotting to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) for 2 h at 100 V. Membranes were blocked for 1 h with 5% nonfat milk in 50 mM Tris, 150 mM NaCl, and 0.05% Tween-20 (TBS-T) at room temperature and subsequently probed with anti-caspase 9 (diluted 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti-cytochrome c (diluted 1:2500; BD Pharmingen, San Jose, CA, USA) overnight. The membranes were rinsed with TBS-T and then incubated with a horseradish peroxidase–conjugated secondary antibody (diluted 1:10,000; Santa Cruz Biotechnology). After the incubation, the membranes were rinsed with TBS-T, and bound antibodies were detected by using enhanced chemiluminescence (Cell Signaling, Danvers, MA, USA) following the manufacturer's instruction. Respective membranes were stripped and probed for anti-β-actin (diluted 1:5000; Sigma) as the loading control. Individual band densities were quantified by ImageJ software.
Statistical analysis
Data were analyzed by one-way analysis of variance using SPSS statistical software (IBM, Somers, NY, USA). Significant differences between two groups were determined by Least Significant Difference testing when P<.05.
Results
Squina DNA enhanced mitochondrial antioxidant status in various tissues of rats
Long-term Squina DNA treatment (18 and 54 mg/kg×14 days) caused dose-dependent increases in mitochondrial GSH levels (16% and 41% [mean values of the five tissues tested], respectively) and GSH/GSSG (29% and 52%), as well as GR (15% and 33%), GPX (15% and 31%), GST (19% and 35%), and ICDH (15% and 38%) activities in various tissues (brain, heart, kidney, liver and skeletal muscle) of rats. Among the different organs, the brain and heart seemed to produce the largest antioxidant response (Fig. 1). Table 1 shows basal values of mitochondrial antioxidant parameters in rats.
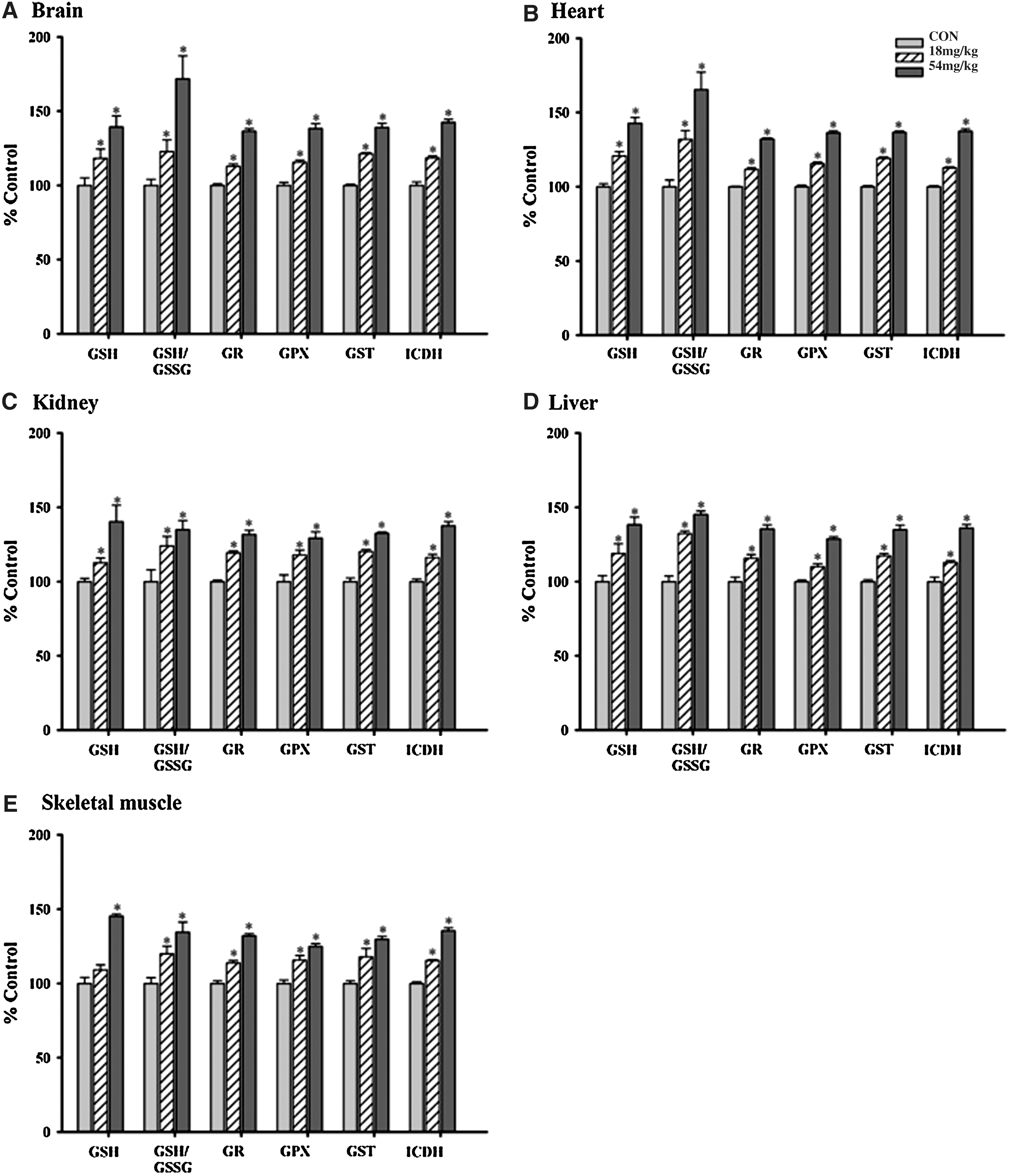
Effects of long-term Squina DNA treatment on mitochondrial antioxidant components in various tissues of rats:
ATP-GC (AUC), ATP generation capacity (area under the curve).
Squina DNA enhanced ATP-GC in various tissues of rats
The enhancement of tissue antioxidant status by Squina DNA was paralleled by dose-dependent increases in the ATP-GC in various tissues (7–26%), with the degree of stimulation being larger in brain and skeletal muscle tissues (Fig. 2).
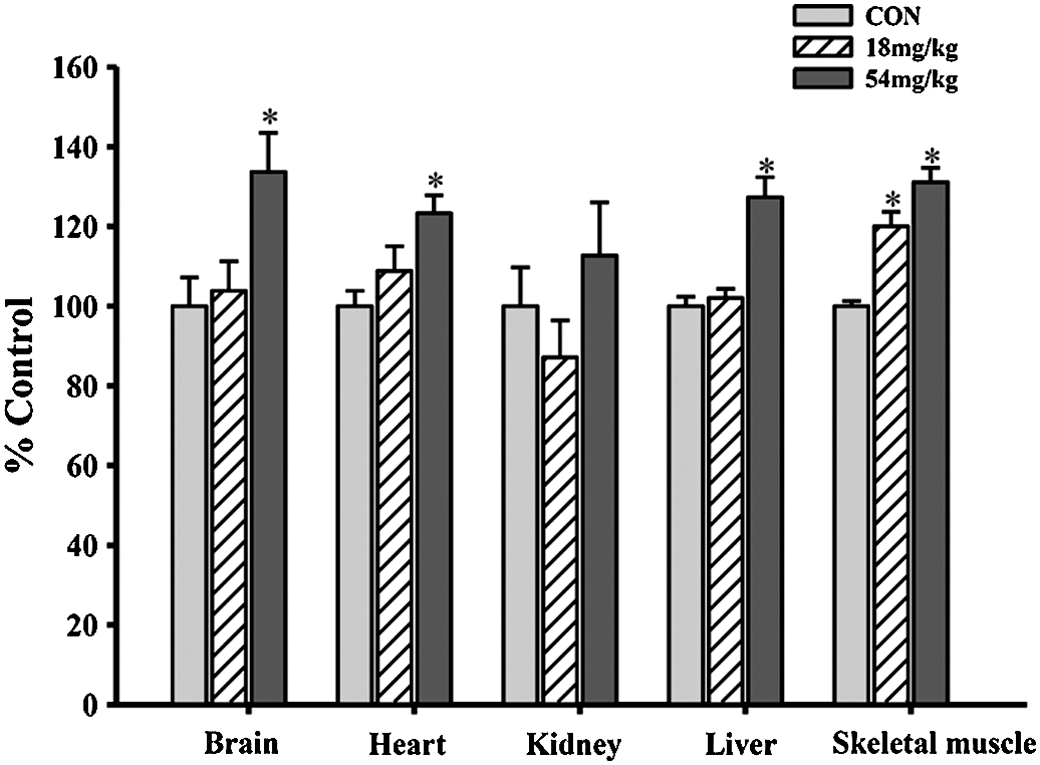
Effects of long-term Squina DNA treatment on mitochondrial ATP-GC in various tissues of rats. Animals were orally administered Squina DNA as described in the legend of Figure 1. ATP-GC was measured using mitochondrial fractions. Data were expressed as percentages of the CON value and are mean±SEM values (n=6). CON values of ATP-GC of various tissues are shown in Table 1. *Significantly different from CON.
Squina DNA protected against cerebral I/R injury in rats
I/R caused cerebral tissue injury, as indicated by the decrease in the amount of viable tissue in brain slices. Although the cerebral blood flow during global cerebral ischemia was not monitored, I/R challenge caused the brain damage in a reproducible manner. Long-term treatment with Squina DNA at daily doses of 18 and 54 mg/kg for 14 days caused dose-dependent protection against cerebral I/R injury in rats, with the degree of protection being 10% and 43%, respectively (Fig. 3).
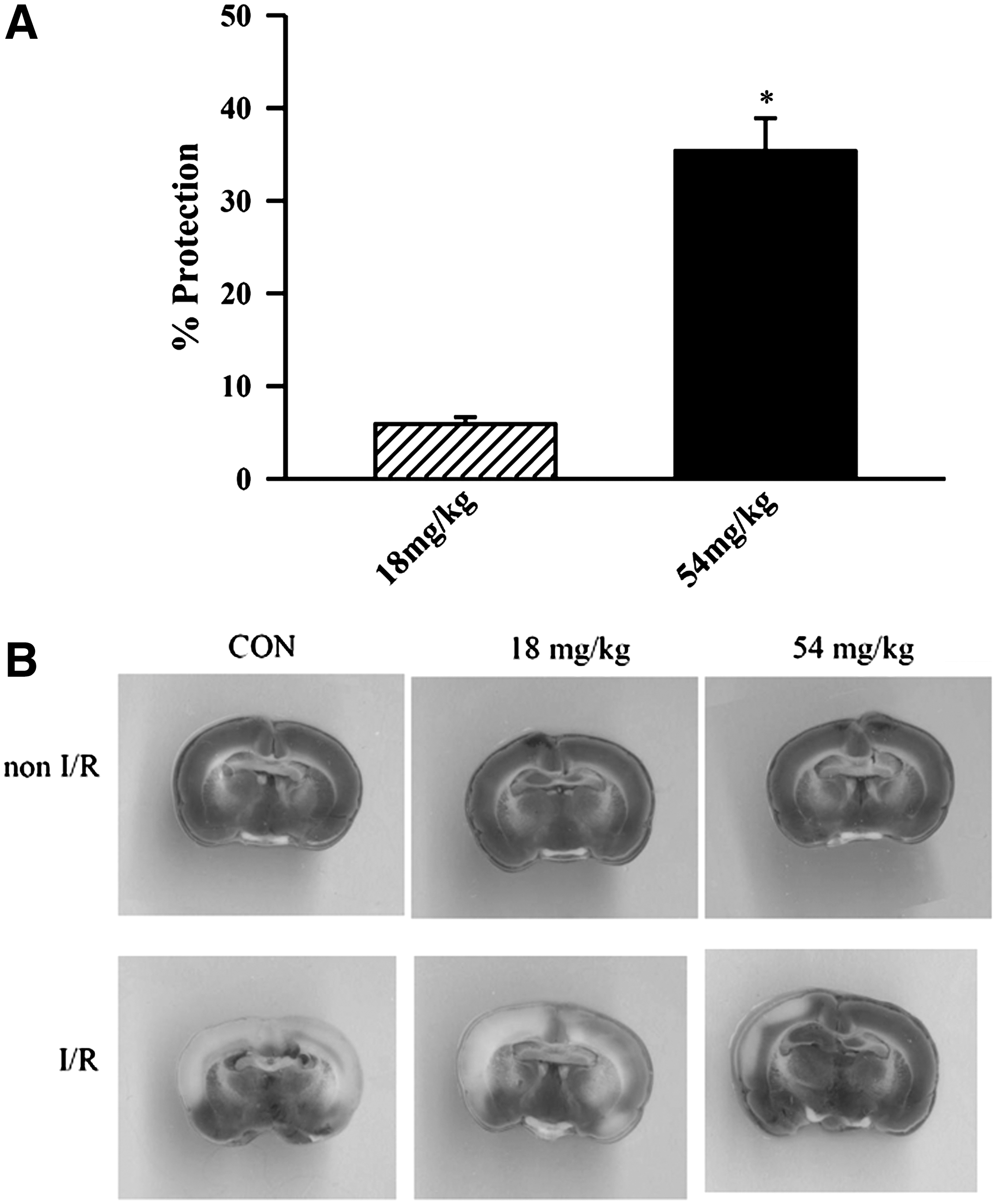
Effect of long-term Squina DNA treatment on cerebral ischemia/reperfusion (I/R) injury in rats. Animals were orally administered Squina DNA, as described in the legend of Figure 1, and then subjected to I/R challenge.
Squina DNA enhanced mitochondrial antioxidant status and membrane integrity in ischemic/reperfused brain tissue in rats
I/R impaired mitochondrial antioxidant status in rat brain tissue, as indicated by the decrease in GSH level (21%) and increase in MDA (40%) production, compared with the non-I/R control (Fig. 4A and B). Squina DNA pretreatment reversed the I/R-induced impairment in mitochondrial antioxidant status in a dose-dependent manner, with GSH and MDA levels being reversed by 31–90% and 28–61%, respectively. I/R disrupted mitochondrial membrane integrity in rat brain tissue, as evidenced by increases in Ca2+ content (61%), sensitivity to Ca2+-induced mitochondrial PT (42%), and cytochrome c release (37%). Squina DNA treatment dose-dependently suppressed the I/R-induced disruption in mitochondrial membrane integrity, with the reversal of mitochondrial parameters by 24–40% for Ca2+ content, 32–73% for Ca2+-induced mitochondrial PT, and 36–91% for cytochrome c release (Fig. 4C–E).
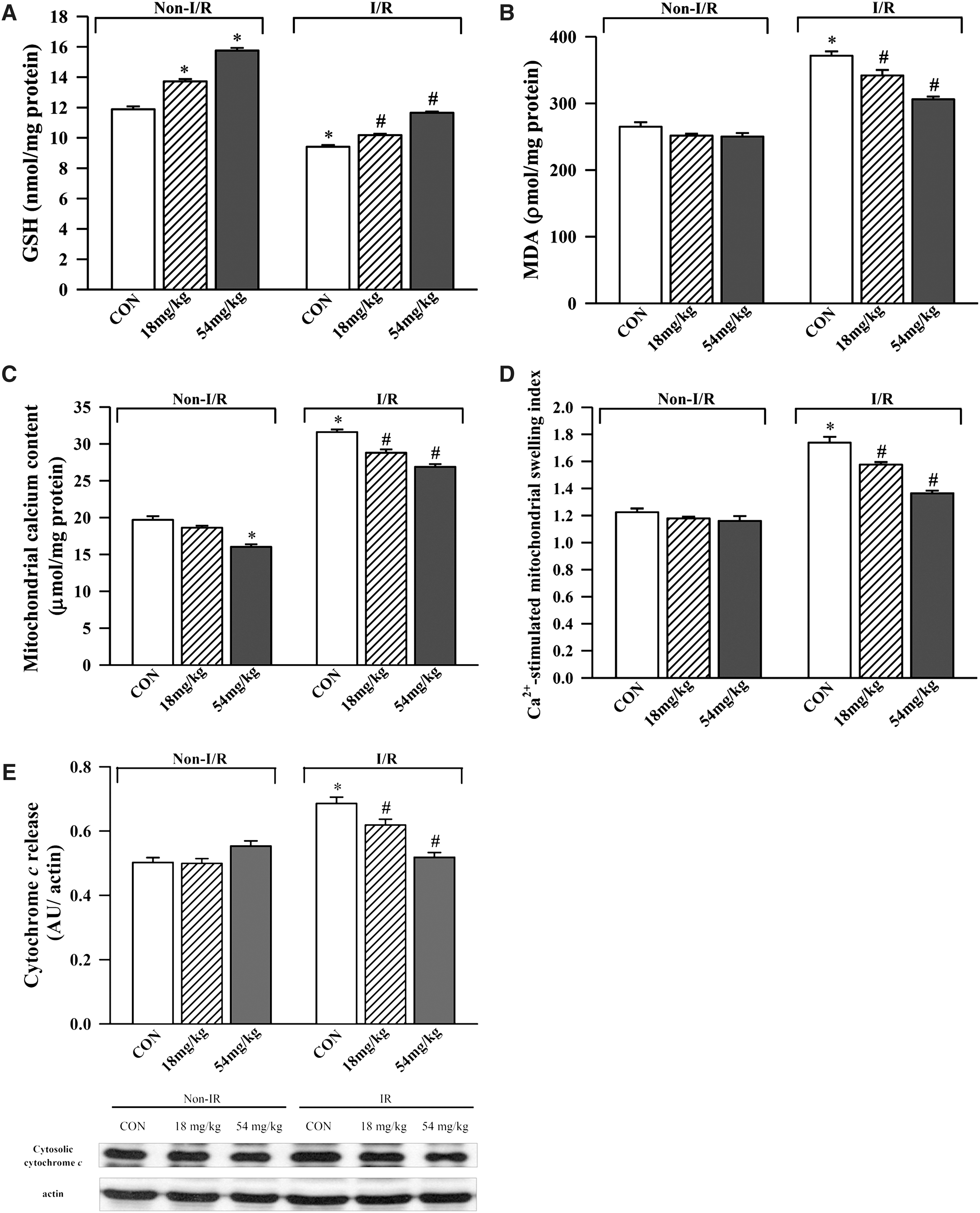
Effect of long-term Squina DNA treatment on mitochondrial antioxidant status and membrane integrity in control and ischemic/reperfused rat brains. Animals were orally administered Squina DNA, as described in the legend of Figure 1, and then subjected to I/R challenge. Mitochondrial antioxidant status was assessed by measurements of
Squina DNA protected against Aβ-induced mitochondrial-dependent cell apoptosis and mitochondrial cytohcrome c release in SH-SY5Y neuronal cells
Aβ challenge (20 and 40 μM) caused apoptosis in SH-SY5Y neuronal cells, as evidenced by 60% and threefold increases, respectively, in the activation of caspase 3 when compared with the unchallenged control. Squina DNA preincubation (150 and 300 μg/mL) dose-dependently decreased the extent of Aβ (40 μM)-induced caspase 3 activation (39.6% and 50.3%, respectively) in SH-SY5Y neuronal cells (Fig. 5A). Aβ challenge (40 μM) caused activation of caspase 9 (42.6%) and increased release of mitochondrial cytochrome c (68.4%), indicative of activation of mitochondrion-dependent cell death pathway as well as disruption in mitochondrial membrane integrity, compared with the unchallenged control. Squina DNA preincubation (300 μg/mL) inhibited caspase 9 activation (by 100%) and cytochrome c release (by 86%) (Fig. 5B and C).
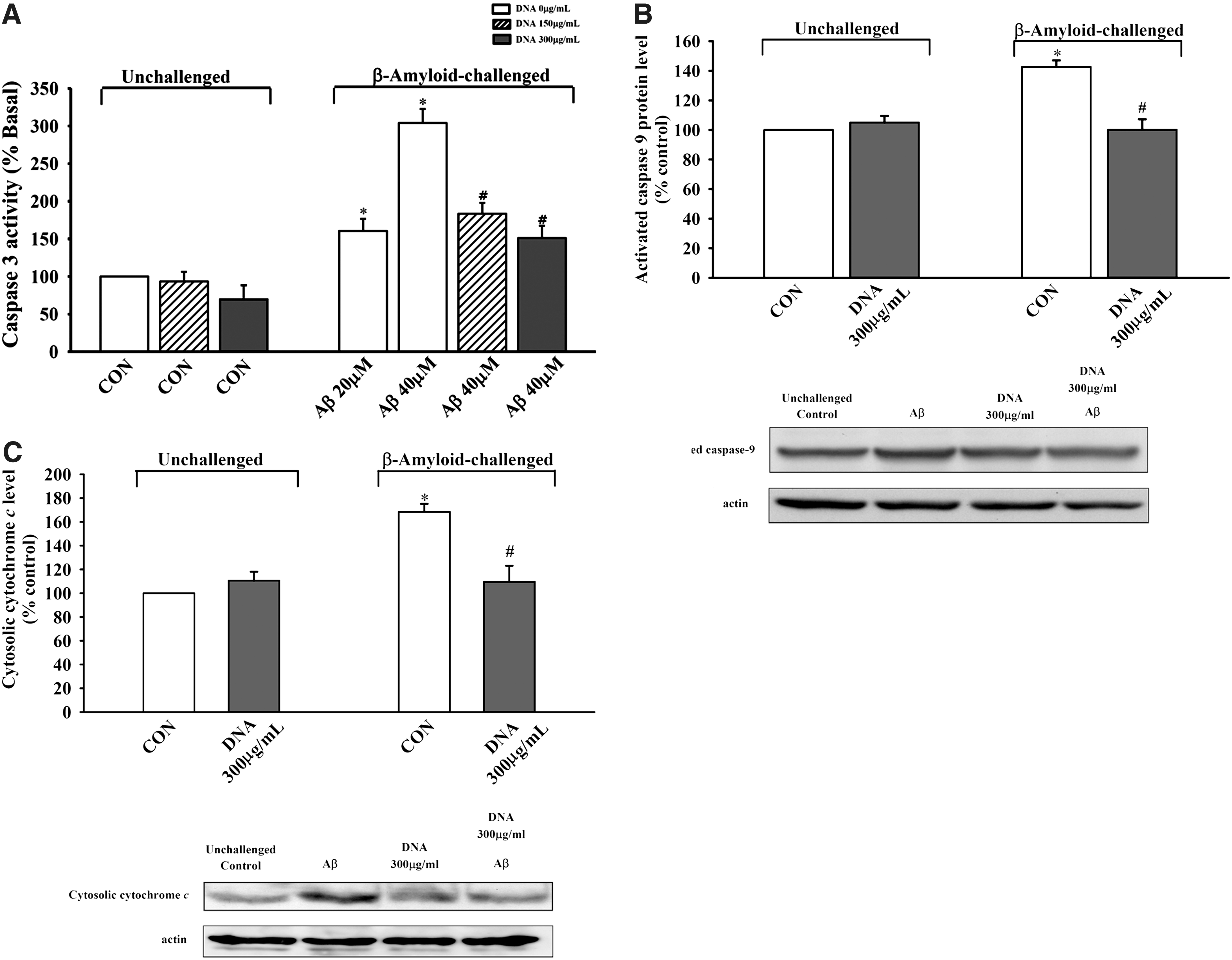
Effect of Squina DNA on β-amyloid (Aβ)-induced mitochondrial-dependent cell apoptosis in SH-SY5Y neuronal cells. SH-SY5Y cells were pretreated with Squina DNA (150 or 300 μg/mL) for 24 h and then subjected to Aβ (20 or 40 μM) challenge for 48 h, as described in Materials and Methods.
Squina DNA elicited a glutathione antioxidant response and induced ROS production in SH-SY5Y neuronal cells
The cytoprotection afforded by Squina DNA was associated with a glutathione antioxidant response. Aβ (40 μM) caused a significant decrease in cellular GSH level (27%), and Squina DNA preincubation (150 and 300 μg/mL) dose-dependently reversed the Aβ-induced GSH depletion (by 57% and 100%, respectively) in SH-SY5Y neuronal cells (Fig. 6). Squina DNA incubation (150 and 300 μg/mL) also caused dose-dependent increases in ROS production (10% and 22%, respectively) in SH-SY5Y neuronal cells (Fig. 7).
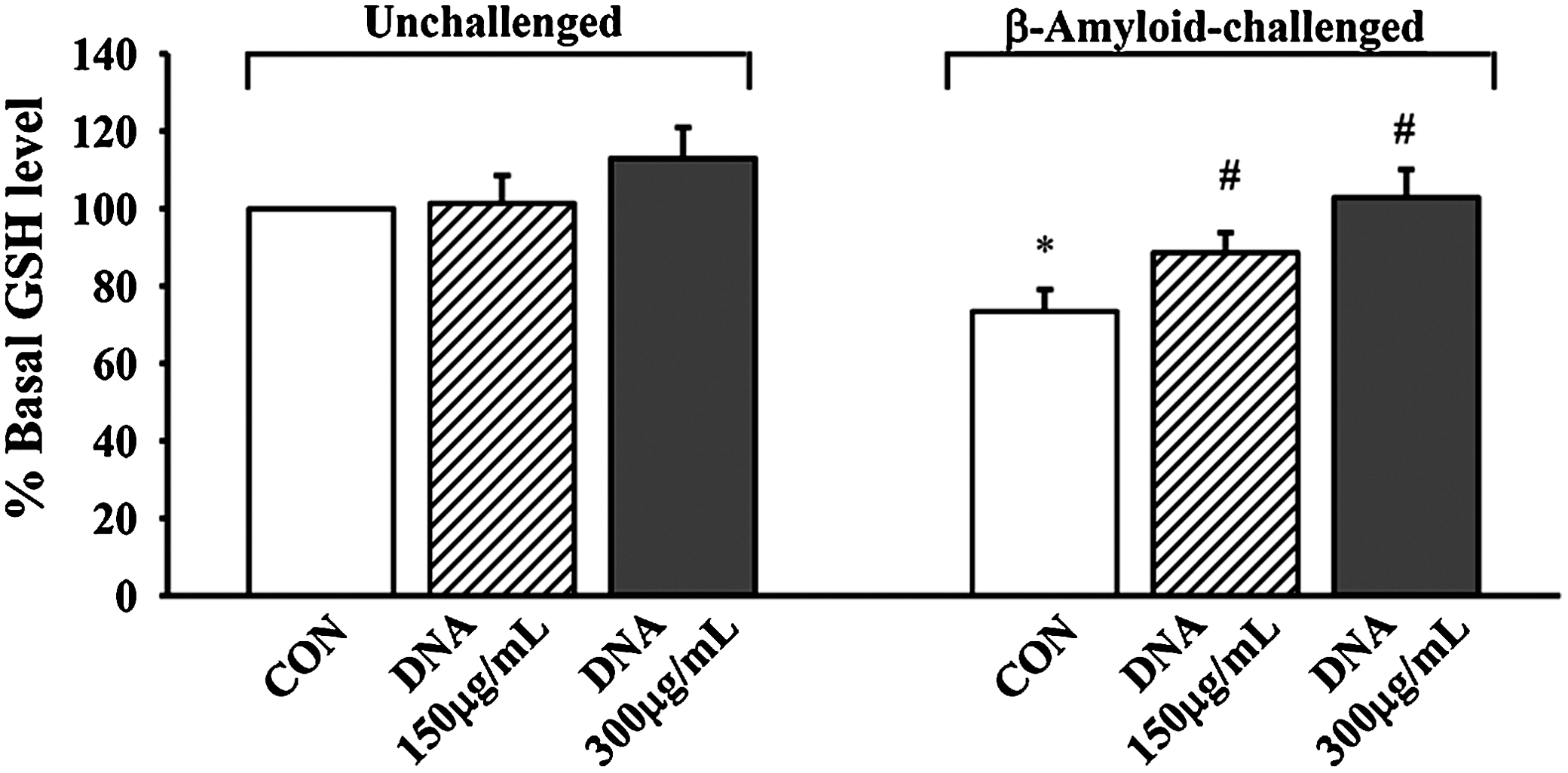
Effect of Squina DNA on cellular GSH levels in CON and Aβ-challenged SH-SY5Y neuronal cells. SH-SY5Y cells were pretreated with Squina DNA ethanol extract (150 or 300 μg/mL) for 24 h and then subjected to Aβ (40 μM) challenge for 48 h. Cellular GSH levels were measured. Data were expressed as percentages with respect to the Squina DNA–untreated and Aβ-unchallenged CON value (15.04±0.37 nmol/mg of protein). Data are mean±SEM values (n≥3). *Significantly different from Squina DNA–untreated and Aβ-unchallenged CON; #significantly different from Squina DNA–untreated and 40 μM Aβ-challenged control.
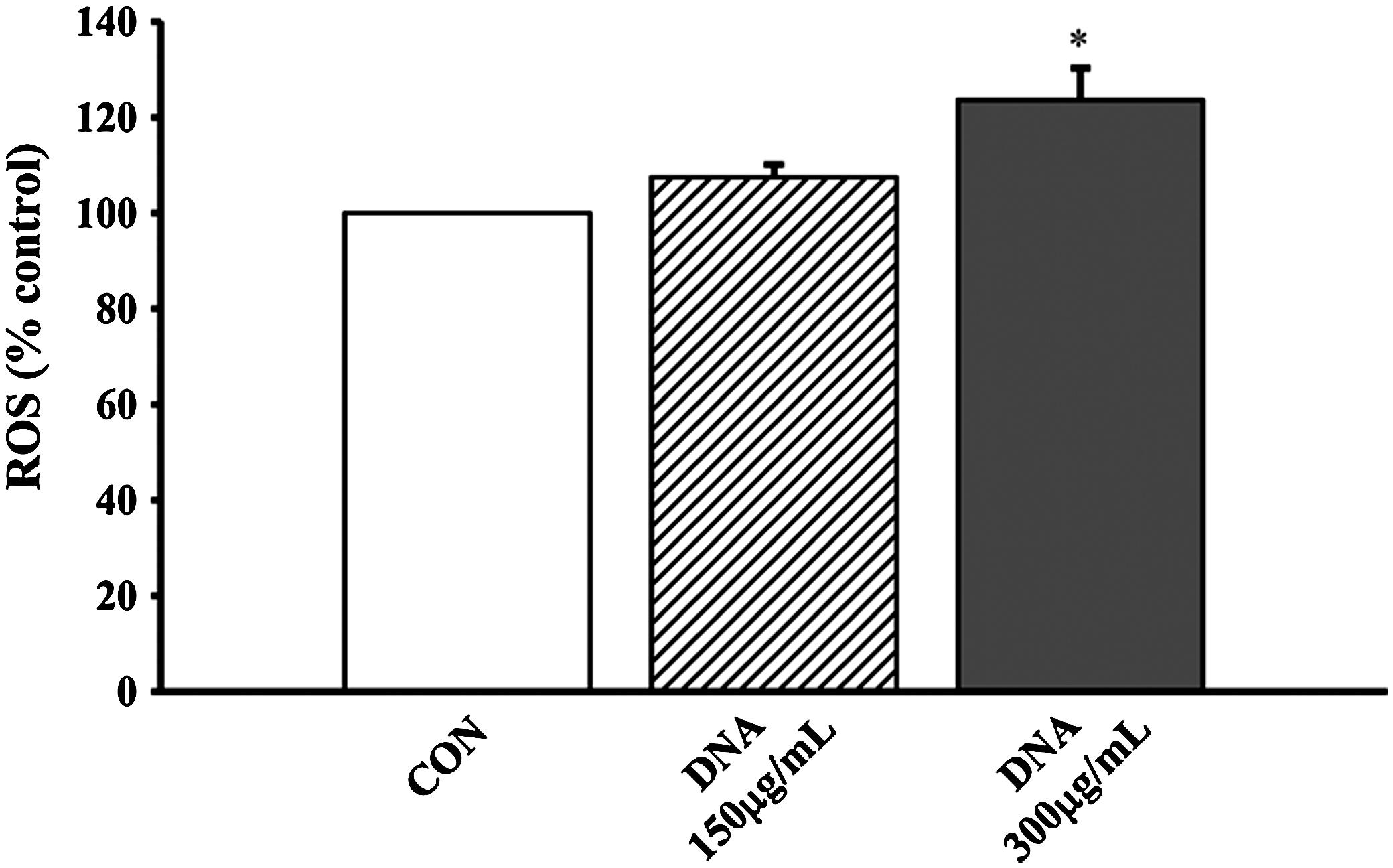
Effect of Squina DNA on reactive oxygen species (ROS) generation in SH-SY5Y neuronal cells. SH-SY5Y cells were treated with Squina DNA ethanol extract (150 and 300 μg/mL), and ROS generation was measured as described in Materials and Methods. Data are mean±SD values (n≥3). *Significantly different from Squina DNA–untreated CON cells.
Discussion
Long-term Squina DNA treatment was found to invariably enhance mitochondrial antioxidant status and functional capacity in various tissues of rats. As mitochondrial antioxidant status and functional capacity are crucial determinants of cell survival and death under oxidative stress conditions, Squina DNA may be used to prevent oxidative stress–induced cell death, which is causally related to the pathogenesis of several diseases such as cardiovascular diseases and neurodegenerative disorders. 18 Oxidative stress occurs when the amount of ROS generated in a system exceeds its capacity to neutralize or to eliminate them, 19 often because of defective production or distribution of endogenous antioxidants or excessive ROS generation. 20 In order to achieve a healthy condition, it is of utmost importance to maintain an efficient antioxidant defense system in the body, particularly in mitochondria.
To cope with the production of ROS during oxidative phosphorylation, mitochondria are equipped with an efficient antioxidant defense system, which is composed of both nonenzymatic and enzymatic antioxidants. Enzymatic antioxidants such as GR, GPX, and GST work cooperatively with nonenzymatic antioxidant GSH to remove the excessive amount of ROS generated from mitochondria. 21,22 The ability of Squina DNA to increase the activity of ICDH, which is responsible for the generation of NADPH, 23 may enhance the GR-catalyzed glutathione redox cycling. This postulation is consistent with the observation that Squina DNA increased the GSH/GSSG ratio in all tested tissues of rats. As an optimal mitochondrial antioxidant status is essential to maintain the functional capacity of mitochondria, the enhancement of mitochondrial ATP-GC by Squina DNA treatment may be an effect secondary to the improvement in mitochondrial antioxidant status.
The enhancement of cerebral mitochondrial antioxidant status by long-term Squina DNA treatment protected against cerebral I/R injury in rats. It is well established that I/R challenge causes neuronal injury through multiple pathophysiological mechanisms, including a burst of ROS production and intracellular/mitochondrial Ca2+ overload, which eventually trigger necrotic and/or apoptotic cell death. 24 Consistent with this, findings obtained from the present study indicated that the cerebroprotection against I/R injury afforded by Squina DNA treatment was paralleled by an amelioration of oxidative stress, as indicated by an increase in mitochondrial GSH level and a decrease in mitochondrial MDA production in I/R-challenged rat brains. The decrease in MDA production, which serves as an indirect index of lipid peroxidation, is presumably a result of the amelioration of oxidative stress caused by the enhancement of GSH level. In addition to oxidative stress, the increased mitochondrial Ca2+ level can trigger the opening of mitochondrial PT pores, a process that leads to apoptotic cell death. 25 Core components of the mitochondrial PT pore putatively include a voltage-dependent anion channel, an adenine nucleotide translocase, and a cyclophilin D displacing peptidyl-prolyl cis–trans isomerase activity. 25 In vivo and in vitro studies showed that high Ca2+ levels can stimulate the opening of mitochondrial PT pores by inducing a conformational change in the adenine nucleotide translocase complex. The resultant formation of solute-nonspecific leaky pores is followed by the release of cytochrome c from mitochondria to cytosol and then a cascade of events leading to apoptosis. 25,26 Consistent with this, mitochondria isolated from ischemic/reperfused rat brain tissue showed an increase in Ca2+ content, increased sensitivity to Ca2+-induced PT, and increased extent of cytochrome c release. Squina DNA treatment, as observed in the present study, was able to ameliorate the I/R-induced impairment in mitochondrial structural integrity.
The cerebroprotection afforded by Squina DNA pretreatment was corroborated by the observation that Squina DNA ethanol extract protected against Aβ-induced apoptosis in SH-SY5Y neuronal cells. Aβ, which is a hydrophobic, nonglycosylated peptide of 39–43 amino acids, forms a β-pleated sheet structure. 27 The aggregated insoluble fragments of Aβ constitute the core substance in senile plaques, a hallmark of postmortem diagnosis of Alzheimer's disease. In vivo and in vitro studies have shown that Aβ is cytotoxic, 28 –31 wherein the aggregation of Aβ imposes a chronic oxidative stress to neuronal cells and eventually induces mitochondria-dependent neuronal cell death. 32,33 In the present study, the Squina DNA ethanol extract effectively prevented the Aβ-induced apoptosis in SH-SY5Y neuronal cells, as indicated by the inhibition of caspase-3 and caspase-9 activation. Aβ-induced cytochrome c release, indicative of mitochondrial PT, was also significantly inhibited by preincubation with the Squina DNA ethanol extract. It is interesting that the incubation with Squina DNA ethanol extract was also found to induce a small amount of ROS production in SH-SY5Y neuronal cells, which may be related to the up-regulation of the glutathione antioxidant system (referred to as glutathione antioxidant response), particularly in Aβ-challenged cells. Although Aβ can induce a relative large extent of ROS production and impose a high level of oxidative stress that is lethal to neuronal cells, 32,33 the modest level of ROS generated by Squina DNA pretreatment was found to induce a cellular glutathione antioxidant response, as indicated by the increase in cellular GSH level. The observation of increased GSH levels in Squina DNA–pretreated and Aβ-challenged cells suggested a lower level of oxidative stress, presumably because of the decrease in Aβ-induced ROS production, in SH-SY5Y neuronal cells. This notion is supported by observations from other studies in which the production of ROS during the cytochrome P-450-mediated metabolism of schisandrin B (an herbal ingredient) was shown to induce cellular glutathione antioxidant response in different types of cell lines. 34,35 It is conceivable that ROS may be generated from the xanthine oxidase–mediated metabolism of purine bases derived from DNA and RNA constituents of Squina DNA, with resultant induction of a glutathione antioxidant response. 36
In conclusion, the results indicate that Squina DNA treatment can enhance mitochondrial antioxidant status, presumably by eliciting a glutathione antioxidant response, and preserve mitochondrial structural integrity, thereby protecting against brain and neuronal injury induced by acute and chronic oxidative stress, respectively.
Footnotes
Author Disclosure Statement
No competing financial interests exist.