
Abstract
3-Nitropropionic acid (3-NP) induces cellular energy deficit and oxidative stress–related neurotoxicity via an irreversible inhibition of mitochondrial complex II enzyme, succinate dehydrogenase. Huntington's disease (HD) is a neurological disorder characterized by cognitive and motor dysfunctions. Lutein is a well-known antioxidant used in the management of oxidative stress related diseases. Clinical trials have supported the beneficial effect of lutein in Alzheimer's disease. The present study was designed to explore possible neuroprotective effects of lutein on 3-NP–induced mitochondrial dysfunction and oxidative stress. Systemic administration of 3-NP (25 mg/kg intraperitoneally [i.p.] for 4 consecutive days) caused loss of body weight and neurobehavioral deficits by hind-limb impairment (Narrow Beam test), motor coordination (locomotor activity) and memory dysfunction (Morris water maze and Elevated Plus maze performance). Biochemical analysis revealed significant increase in lipid peroxidation, nitrite concentration, reduced gutathione levels, and acetyl cholinesterase levels and depleted catalase activities in rat brain. The activities of mitochondrial complexes (I, II, IV, and MTT assay) were found to be significantly lowered in brain mitochondria. Daily lutein (50 or 100 mg/kg orally [p.o.]) administration for 14 days significantly improved body weight, neurobehavioral alterations and attenuated oxidative stress and improved mitochondrial enzymes complex activities of rat brain. Histopathological examination further affirmed the neuroprotective effect of lutein on 3-NP induced pathological lesions. The present study indicates that lutein is a promising candidate for the management of HD and related conditions.
Introduction
H
3-Nitropropionic acid (3-NP), a mycotoxin produced by the fungus Arthrinium sp., is a suicide inhibitor of respiratory chain and Krebs cycle enzyme succinate dehydrogenase (SDH). 3-NP interferes with ATP synthesis in brain mitochondria, leading to mitochondrial dysfunction, and also produces selective striatal lesions and induces proliferative changes in the dendrites of spiny neurons, resulting in neuronal degeneration within basal ganglia and movement dysfunction as observed in HD patients. 5 3-NP depletes antioxidant defense enzymes and increases levels of reactive oxygen/nitrogen species in different areas of the brain, leading to oxidative damage. 6
Recent studies have shown that the antioxidants of plant origin with free-radical scavenging properties can function as therapeutic agents in several diseases caused by oxidative stress. 7 As a result, an increasing number of researchers have been studying the effects of antioxidants in the central nervous system to develop new therapies to treat these conditions. Lutein is a xanthophyll and one of 600 known naturally-occurring carotenoids which are synthesized only by plants. Lutein is showing antioxidant properties 8 which may be attributed to its unique chemical structure; it not only has conjugated double bonds but also has two hydroxyl groups on both ends making it a stronger antioxidant compared to other carotenoids. 9 Lutein has been studied for amelioration of conditions such as diabetic neuropathy and age-related macular degeneration (AMD), which are mainly associated with oxidative stress. 10 Astaxenthin, a stereoisomer of lutein, has been studied for its beneficial effects in apoptosis. 11 Clinical trials also supported the beneficial effect of lutein in Alzheimer's disease. 12 The present study was designed to elucidate the beneficial effects of lutein as an antioxidant in ameliorating neurobehavioral deficits, mitochondrial dysfunction, and oxidative stress by 3-NP–induced neurotoxicity in an animal model of HD.
Materials and Methods
Chemicals
All of the chemicals used in the present study were of analytical grade and were purchased from Sigma Chemicals Co., Sisco Research Laboratories, and SD Fine Chemicals. Lutein was supplied by OmniActive Health Technologies, Ltd., Mumbai, India.
Animals and treatment schedule
Female Sprague-Dawley rats (200–250 g) were used in the study. Animals were acclimatized to laboratory conditions before experimentation. The animals were kept in groups of four per cage under standard conditions of light–dark cycle, with food and water ad libitum in acrylic box cages with soft bedding. All of the experiments were carried out between 9:00 a.m. and 3:00 p.m. The protocol followed was approved by the Institutional Animal Ethics Committee and was in accordance with guidelines of Committee for the Purpose of Control and Supervision of Experiments on Animals for use and care of laboratory animals.
3-NP (Sigma Chemical) was diluted with saline (adjusted to pH 7.4) and administered intraperitonealy to rats. Lutein was suspended in 0.5% sodium carboxy-methyl cellulose solution and administered orally in a constant volume of 0.5 mL/200 g of body weight. Animals were randomly divided into six groups of eight animals.
Group I received vehicle for lutein (p.o.) and also normal saline (i.p.) for 14 days. Group II received vehicle for 14 days and 3-NP (25 mg/kg i.p.) for the last 4 days (days 11–14). Groups III and IV received lutein (50 or 100 mg/kg p.o., respectively) for 14 days. Groups V and VI received lutein (50 or 100 mg/kg, p.o., respectively) for 14 days and 3-NP (25 mg/kg i.p.) for the last 4 days (days 11–14). 3-NP was administered 1 h after lutein administration.
Measurement of body weight
Body weights of animals were recorded on the first and last day of the experiment. Percent change in body weight was calculated as change in body weight (initial weight minus final weight) divided by initial body weight, multiplied by 100%.
Behavioral assessments
All animals were trained for 8 days before starting the experiment.
Narrow Beam test
The Narrow Beam test was used to measure hind-limb impairment as described previously. 13 The narrow beam used for the present experiments was a 105 cm long and 4 cm broad wooden beam. The beam was suspended 80 cm from the ground by wooden supports at either end. At the start end of the beam, a line was drawn 20 cm from the end of the beam. During the test, the rat was placed entirely within this 20 cm starting zone facing its home cage and a stopwatch started immediately upon release of the animal. The time taken to traverse the beam was recorded. The maximum time allowed for the task was 2 min; a fall was also recorded as a maximum time. The test was performed on days 0 and 15 of the study.
Elevated Plus maze
The Elevated Plus maze was used to evaluate spatial long-term memory according to the method of Lee O-H et al. 11 Briefly, the apparatus consisted of two open arms and two closed arms. The arms extended from a central platform, and the maze was elevated to a height of 50 cm from the floor. On the first day, each animal was placed at the end of an open arm. Transfer latency (TL) was recorded as the time taken by the rat to move into one of the enclosed arms. If the animal did not enter a closed arm within 90 s it was gently pushed into one closed arm and the TL latency was assigned as 90 s. The rats were allowed to explore the maze for 20 s, and then return to the home cage. TL was recorded on days 0 and 15 of the study.
Morris water maze test
The Morris water maze test was performed for assessment of cognitive performance by discrete modification to the method of Kumar et al. 14 The maze consists of a circular plastic tank (160 cm in diameter and 35 cm in height). The tank was divided by four fixed points on its perimeter into four quadrants. Water was made opaque by adding milk. It contains an escape platform of the same color as the rest of the basin (to eliminate any false positive results due to vision) placed in the same quadrant of the basin throughout the trials, and was placed 2 cm below the water surface. Rats were placed gently at a start point in the middle of the rim of a quadrant not containing the escape area with their face to the wall. If the rat did not escape on to the platform within 90 s it was guided to the platform and allowed to stay on it for 20 s. Animals received four trials per day separated by 10 min for 5 successive days (acquisition trials) during which the time required to reach the platform was calculated. After the acquisition trials, time taken to find the hidden platform was recorded on day 0 and the 15th day after start of drug administration.
Assessment of gross behavioral activity (locomotor activity)
The locomotor activity was monitored using an actophotometer on the first and last days of study. Each interruption of a beam on the x- or y-axis generated an electric impulse, which was presented on a digital counter. The apparatus was placed in a darkened, light- and sound-attenuated, and ventilated testing room. Each animal was observed over a period of 5 min and values are expressed as counts per 5 min. 15
Mitochondrial respiratory chain enzymes activities
Animals were sacrificed using a carbon dioxide chamber on day 15. Mitochondria were isolated by a modified method of Sandhir et al. 16 Briefly, the whole brain was dissected, rinsed in ice-cold isotonic saline, and homogenized with 10 volumes (w/v) of ice-cold extraction buffer (10 mM Tris-HCl, pH 7.4, 0.44 M sucrose, 10 mM ethylenediaminetetraacetic acid [EDTA], and 0.1% bovine serum albumin [BSA]). The homogenate was centrifuged using a Remi cooling centrifugor (RH 106) at 5000 rpm for 30 min at 4°C. The pellet was discarded and the supernatant recentrifuged at 10,000 rpm for 30 min at 4°C. Supernatant obtained before recentrifugation was used for the estimation of acetyl cholinesterase (AChE) and nitrite levels and oxidative stress parameters. After recentrifugation, the crude mitochondrial pellet was separated, washed with extraction buffer and centrifuged at 10,000 rpm for 30 min at 4°C. The final mitochondrial pellet was resuspended in buffer containing 0.44 M sucrose in 10 mM Tris-HCl, pH 7.4. Suspensions of the mitochondrial pellet were used for the following mitochondrial enzymes estimations.
Nicotinamide adenine dinucleotide (NADH) dehydrogenase activity was measured by the method of King and Howard, 17 which involves catalytic oxidation of NADH with subsequent reduction of cytochrome c. Briefly, mitochondrial fraction (0.05 mL) was added to 3.0 mL of reaction mixture, which contained 0.35 mL (0.2 M) glycyl glycine pH 8.5, 0.10 mL (6 mM) NADH, 0.1 mL (1 mM) oxidized cytochrome c and 0.05 mL (0.02 M) NaHCO3 and 2.40 mL water. The increase in absorbance was read at 550 nm for 3 min. Results were expressed as nmol NADH oxidized/min/mg protein.
SDH activity in the mitochondrial fraction was estimated according to the method of King. 18 The reaction mixture consisted of 1.5 mL of phosphate buffer (0.2 M, pH 7.8), 0.2 mL of succinic acid (0.6 M, pH 7.8), 0.3 mL of BSA (1%, w/v), 0.1 mL of 0.03 M potassium ferricyanide and the reaction was started by addition of mitochondrial fraction (0.05 mL). The decrease in absorbance was recorded at 420 nm for 3 min. Results were expressed as nmol succinate oxidized/min/mg protein.
Cytochrome oxidase was assayed in the mitochondrial fraction according to the method of Sottocasa et al. 19 Briefly, cytochrome c (3 mM) was reduced first by the addition of a few crystals of sodium borohydride, and then neutralized to pH 7.0 by 0.1 M HCl. Reduced cytochrome c (0.3 mM) was added to 0.075 M phosphate buffer (pH 7.4), and the reaction was initiated by addition of mitochondrial suspension (50 μL). The decrease in absorbance was measured at 550 nm for 3 min. Results were expressed as nmol cytochrome c oxidized/min/mg protein, using molar extinction coefficient of cytochrome c (19.6/mM/cm).
MTT reduction was used to assess the mitochondrial function by the method described by Kamboj et al. 20 The reaction mixture containing mitochondrial fraction (500 μL) and MTT (500 μL, 0.1 mg/mL) was incubated at 37°C for 120 min, and then centrifuged to obtain formazan pellet. The pellets were dissolved in 1 mL of absolute ethanol and the mixture was recentrifuged. The absorbance of the supernatant was measured at 595 nm. Results were expressed as mg formazan formed/min/mg protein.
Mitochondrial oxidative stress parameters
The amount of malondialdehyde (MDA), a measure of lipid peroxidation was quantified by reaction with thiobarbituric acid (TBA) according to the method described by Ohkawa et al. 21 The tissue supernatant (0.1 mL) was mixed with 2.5 mL TBA (0.7% in 30% glacial acetic acid). The mixture was incubated at 95°C for 60 min; the mixture was cooled under tap water. After cooling, the mixture was centrifuged at 4000 rpm for 10 min. The absorbance of the supernatant was read at 532 nm. The MDA content of the test tissue was calculated from the standard graph and results were expressed as nmol MDA/mg protein.
Catalase activity was assayed by the method of Luck 22 wherein the breakdown of hydrogen peroxide (H2O2) is measured at 240 nm. Briefly, assay mixture consists of 3 mL of H2O2 phosphate buffer and 0.05 mL of tissue supernatant and change in absorbance was recorded at 240 nm for 3 min. The results were expressed as micromole H2O2 decomposed per milligram of protein/min.
Reduced glutathione was estimated according to the method described by Ellman. 23 To 0.2 mL of tissue supernatant, 0.6 mL of 0.2 M Tris-EDTA, 50 μL of 0.01 M 5,5′-dithiobsis(2-nitrobenzoic acid) (DTNB), and 2.5 mL methanol was added. The mixture was incubated at 37°C for 30 min with occasional shaking. The mixture was then centrifuged at 4000 rpm for 15 min. The supernatant was separated. The intensity of color developed was determined at 412 nm against blank treated in the same way, replacing tissue supernatant with distilled water. The values of reduced glutathione (GSH) were obtained by interpolation of a standard plot of glutathione and values were expressed as reduced glutathione μmole/mg protein.
Nitrite levels were determined by a colorimetric assay using Greiss reagent (0.1% naphthylethylene diamine dihydrochloric acid and 1% sulfanilamide in 5% phosphoric acid). Equal volumes (500 μL) of tissue supernatant and Greiss reagent were mixed, the mixture was incubated for 10 min at room temperature in dark and the absorbance was determined at 540 nm. 24 The concentration of nitrite in supernatant was determined from a sodium nitrite standard and expressed as μmole/mg protein.
AChE is a marker of loss of cholinergic neurons in the forebrain. The AChE activity was assessed by Ellman method. 25 The assay mixture contained 0.05 mL of tissue supernatant, 3 mL of sodium phosphate buffer (pH 8), 01 mL of acetyl-thiocholine iodide and 0.1 mL DTNB. The change in absorbance was measured for 2 min at 412 nm using a Perkin-Elmer Lambda 20 spectrophotometer. The results were expressed as μmole acetyl-thiocholine iodide hydrolyzed/min/mg protein.
The protein content was estimated according to the method of Lowry et al. 26 One hundred microliters of supernatant was added to 2 mL of Lowry reagent and incubated for 10 min at room temperature. Later, 0.2 mL Folins ciocalteaus reagent was added, mixed immediately and incubated for 30 min at room temp. Absorbance was measured at 660 nm, and results were expressed as mg/mL using BSA as standard.
Histopathological examination
Brains were fixed in 10% formalin solution for 24 h using the Hartz technique. 27 The fixed tissues were washed in tap water, dehydrated in a series of alcohol, cleared in xylene, then embedded in paraffin blocks. Sections 5 μm thick were obtained from the blocks and stained by hematoxylin and eosin. The tissue sections were then examined under a light microscope at different magnifications and photographs were taken.
Statistical analysis
All values are expressed as mean±standard deviation of eight animals per group. Data were analyzed using one way analysis of variance followed by Tukey's test. Values with P<.05 were considered as statistically significant.
Results
Effect of lutein on body weight in 3-NP–treated rats
3-NP treatment caused significant decrease in body weight on day 15 as compared to the vehicle-treated group. Further lutein treatment (50 or 100 mg/kg) attenuated the body weight of 3-NP–treated rats compared to the group treated with only 3-NP. There was no significant change in the initial and final body weights of vehicle- and per se–treated animals (Fig. 1).
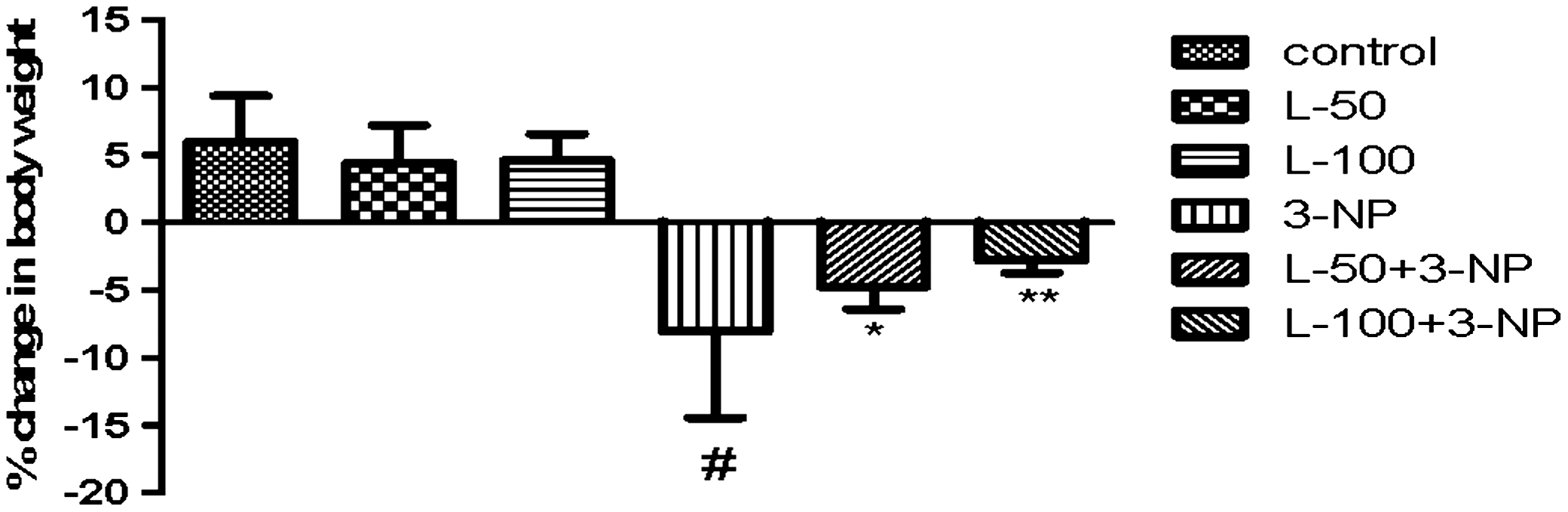
Effect of lutein on body weight in 3-nitropropionic acid (3-NP)–treated rats. Values are expressed as mean±standard deviation (SD); n=8. Significance was determined using one-way ANOVA followed by Tukey's test. #Significantly different from control group (P<.05). *Significantly different from 3-NP–treated group (P<.05). **Significantly different from L-50+3-NP–treated group. ANOVA, analysis of variance; L-50, lutein 50 mg/kg.
Effect of lutein on neurobehavioral deficits
Hind limb impairment was measured by the Narrow Beam test in terms of time taken by each animal to transverse the beam (Fig. 2). On day 0, time taken by the animals ranged between 3 and 10 s in all groups. On day 15, the time taken significantly increased in 3-NP treated animals as compared to the control group. 3-NP–challenged rats pretreated with lutein (50 or 100 mg/kg) showed a significant decrease in time compared to 3-NP treatment alone. Per se treatment of lutein (50 or 100 mg/kg) did not show any significant change compared with 3-NP treatment.

Effect of lutein administration in 3-NP–treated rats on the Narrow Beam test in terms of total time. Values are expressed as mean±SD; n=8. Significance was determined using one-way ANOVA followed by Tukey's test. #Significantly different from control group (P<.05). *Significantly different from 3-NP treated group (P<.05). **Significantly different from L-50+3-NP–treated group.
Spatial memory was assessed in terms of TL in an Elevated Plus maze task. 3-NP treatment significantly delayed TL as compared to the vehicle-treated group. In contrast, pretreatment with lutein (50 or 100 mg/kg) to 3-NP–challenged rats shortened TLs compared to 3-NP treatment. However, treatment with lutein only (50 or 100 mg/kg) did not show any significant change in TL as compared to the vehicle-treated group (Fig. 3).

Effect of lutein administration on transfer latency in the Elevated Plus maze task in 3-NP–treated rats. Values are expressed as mean±SD; n=8. Significance was determined using one-way ANOVA followed by Tukey's test. #Significantly different from control group (P<.05). *Significantly different from 3-NP–treated group (P<.05). **Significantly different from L-50+3-NP–treated group.
Cognitive impairment was assessed using the Morris water maze test in terms of time required by animals to locate the platform (escape latency). On day 15, escape latency was significantly increased in the 3-NP–treated group as compared to the control group. Pretreatment with lutein (50 or 100 mg/kg p.o.) showed a significant improvement in escape latency compared with the 3-NP–treated group. Treatment with lutein only (50 or 100 mg/kg) did not affect escape latency time in comparison with control treatment (Fig. 4).

Effect of lutein administration on cognitive impairment in the Morris water maze in 3-NP–treated rats. Values are expressed as mean±SD; n=8. Significance was determined using one-way ANOVA followed by Tukey's test. #Significantly different from control group (P<.05). *Significantly different from 3-NP–treated group (P<.05). **Significantly different from L-50+3-NP–treated group.
3-NP treatment caused a significant decrease in locomotor activity as compared to the vehicle-treated group. In contrast, treatment with lutein (50 or 100 mg/kg) significantly improved locomotor activity in 3-NP–treated rats compared with the animals treated with 3-NP only. Per se treatment of lutein (50 or 100 mg/kg) did not show any significant change (Fig. 5).
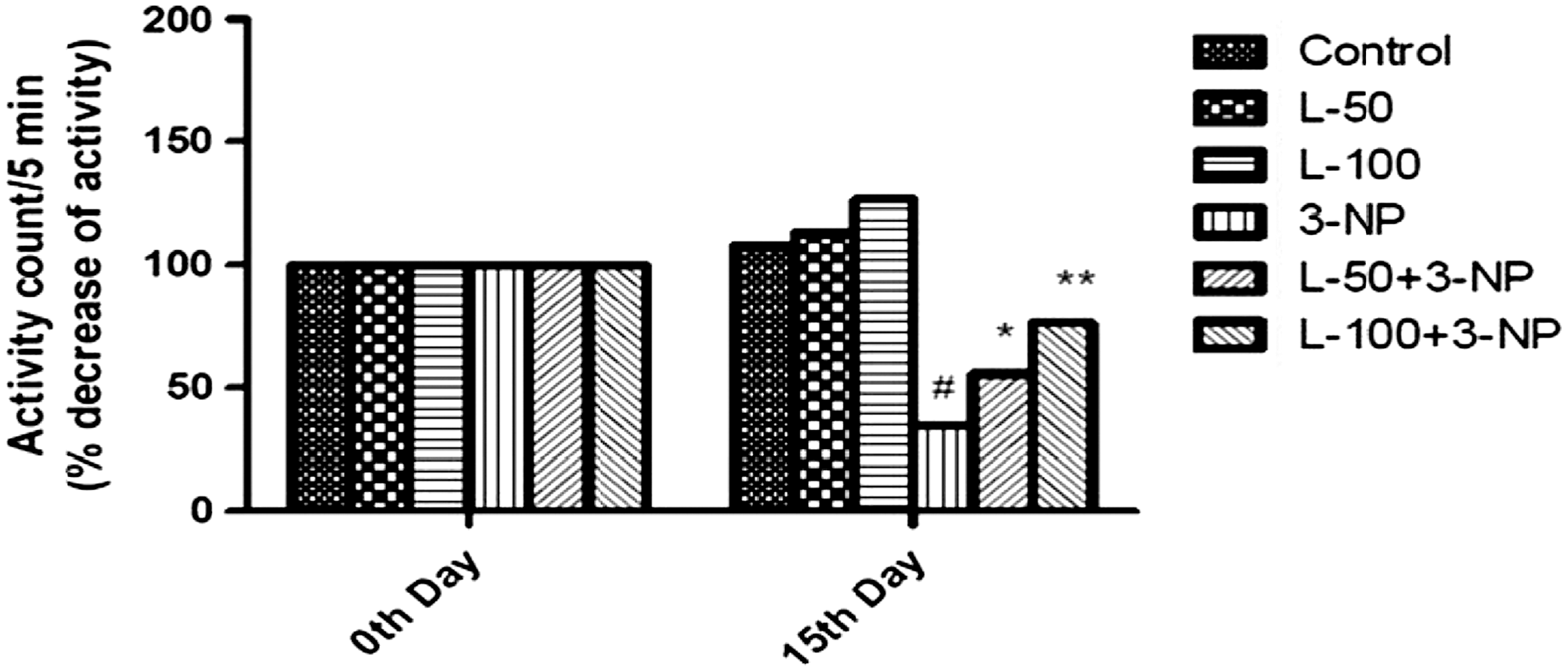
Effect of lutein administration on locomotor activity in 3-NP–treated rats. Values are expressed as mean±SD; n=8. Significance was determined using one-way ANOVA followed by Tukey's test. #Significantly different from control group (P<.05).*Significantly different from 3-NP–treated group (P<.05). **Significantly different from L-50+3-NP–treated group.
Effect of lutein on mitochondrial respiratory chain enzymes in 3-NP–treated rats
The activities of various mitochondrial ETC enzymes are presented in Table 1.
Values are expressed as mean±SD; n=8 (one-way ANOVA followed by Tukey's test).
Groups were treated as follows: control, normal vehicle treated rats; L-50, lutein (50 mg/kg); L-100, lutein (100 mg/kg); 3-NP, 3-nitropropionic acid (25 mg/kg).
Significantly different from control group (P<.05).
Significantly different from 3-NP group (P<.05).
Significantly different from (lutein 50 mg/kg+3-NP) and 3-NP treated group (P<.05).
SDH, succinate dehydrogenase; SD, standard deviation; ANOVA, analysis of variance.
Mitochondrial dysfunction induced by 3-NP exhibited significant (P<.05) decrease in activities of mitochondrial enzyme complexes (I, II, IV, and MTT activities) as compared to the vehicle-treated group. Treatment with lutein (50 or 100 mg/kg) for 14 days dose-dependently counteracted the deleterious effects of 3-NP by increasing the mitochondrial enzyme complexes' activities as compared to 3-NP treatment. However, treatment with lutein only (50 or 100 mg/kg) attenuated complex I (NADH dehydrogenase) activity compared with vehicle treatment, but the activities of other enzymes (II, IV, and MTT reduction) were not altered significantly.
Effect of lutein on brain lipid peroxidation, antioxidant enzyme (catalase), and GSH levels
3-NP–treated rats exhibited a significant decrease in activities of catalase and reduced GSH as compared to controls. Pretreatment with lutein (50 or 100 mg/kg) dose-dependently counteracted the deleterious effects of 3-NP by increasing the levels of antioxidant parameters as compared to 3-NP treatment. Antioxidant enzyme (Catalase) activity increased significantly after the per se treatment of lutein (50 or 100 mg/kg) compared with control treatment, whereas levels of reduced GSH were not affected (Table 2).
Values are expressed as mean±SD; n=8 (one-way ANOVA followed by Tukey's test).
Significantly different from control group (P<.05).
Significantly different from 3-NP group (P<.05).
Significantly different from (lutein 50 mg/kg+3-NP) and 3-NP treated group (P<.05).
There was a significant elevation in the level of liquid peroxidation in 3-NP–treated rats compared with vehicle-treated rats. However, treatment with lutein (50 or 100 mg/kg) for 14 days to 3-NP–treated rats significantly decreased these levels dose-dependently, compared to 3-NP–treated rats. Per se treatment of lutein (50 or 100 mg/kg) did not affect TBARS levels in comparison with vehicle treated group (Table 2).
Effect of lutein on nitrite and AChE enzyme levels
As shown in Table 2, systemic administration of 3-NP caused significant increases in levels of nitrite and AChE as compared to the control group. However, lutein pretreatment (50 or 100 mg/kg) to 3-NP–challenged rats significantly decreased the elevated levels of nitrite and AChE compared with 3-NP treatment alone. Per se treatment of lutein (50 or 100 mg/kg) did not alter these levels as compared to control group.
Effect of lutein on histological alterations in striatum
Brain tissue sections from rats of the control group showed normal histological structure (Fig. 6). 3-NP treatment produced severe degeneration of neurons with complete loss of cell detail and architecture, and with the appearance of basophilic nuclear remnants in the central zone. Animals pretreated with lutein before 3-NP administration showed mild focal gliosis associated with swelling in the endothelial lining of blood capillaries. Striata of lutein-only treated rats showed no histopathological alterations compared to control animals.
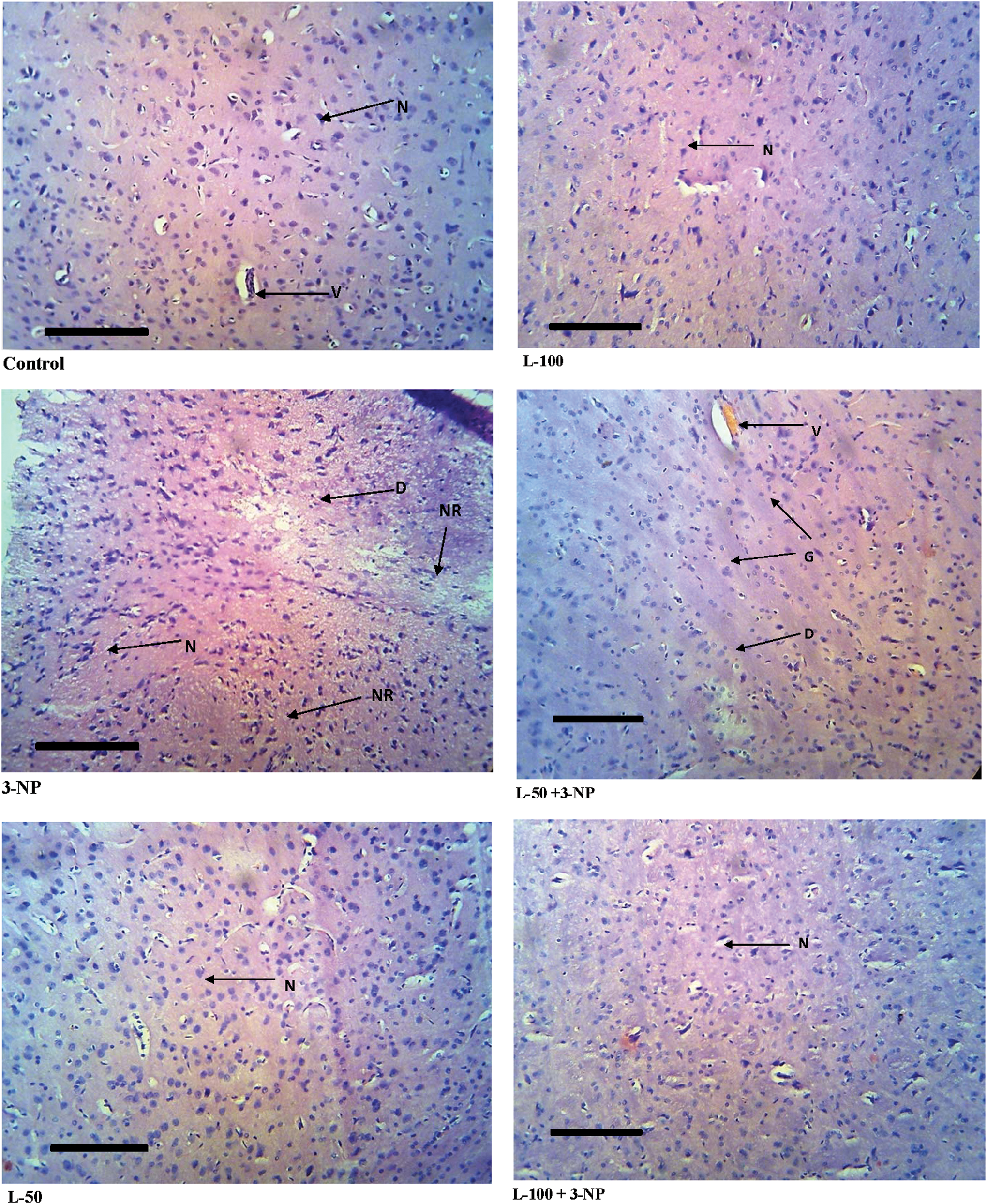
Histopathological pictures from the sections through the striatum of the brain, showing the effects of lutein on 3-NP–induced pathological lesions. Color images available online at
Discussion
This is the first extensive report of the effect of lutein in 3-NP–induced HD-like symptoms in rats. Lutein is a xanthophyll and has antioxidant properties. 8 The pathogenesis of HD is not yet fully understood, but studies on 3-NP–induced neurotoxicity provide good insight into its pathology and clearly indicate the involvement of oxidative stress and mitochondrial dysfunction. Consequently, pharmacological inhibitors of mitochondrial complex II (3-NP) have been found to induce striatal damage and motor phenotypes in animals, which closely resembles the symptoms seen in HD patients. 4 HD-like symptoms include chorea, memory impairment, oxidative stress, and mitochondrial dysfunction in the rat brain. 1
3-NP is a suicide inhibitor of respiratory chain and Krebs cycle enzyme SDH. Inhibition of these enzymes in the ETC leads to an increase in electron leakage from mitochondria and production of reactive oxidative species (ROS), which causes oxidative stress.
28
Roles of various antioxidants have been recently reported in the management of HD-like symptons: lycopene and epigallocatechin-3-gallate,
29
Withania somnifera root extract,
30
flavonoid kameferol,
31
hesperidin and naringenin,
32
sartraline,
33
S-allylcysteine,
34
Antioxidants are molecules that act against any form of oxidative stress and its associated ill effects on cellular systems. They neutralize ROS and other kinds of free radicals produced as a consequence of oxidative stress and have attracted the attention of clinicians due to therapeutic potential. 5 Lutein, a xanthophyll having antioxidant properties, 8 shows beneficial effects in AMD, cataracts, cancer, coronary heart disease, and stroke, 3 and is under investigation for treatment of Alzheimer's disease. 12 Supporting the present investigation, lutein is capable is terminating free radical reactions and protecting our body from oxidative stress, which is the basis for neurodegenerative disease. In our experiment, we found that lutein protected the brain from 3-NP–induced oxidative stress and mitochondrial dysfunction via favorably improved neurobehavioral, biochemical, and histopathological parameters, suggesting its neuroprotective action.
HD patients often show gradual decreases in body weight despite normal and increased energy intakes. Decrease in body weight can be considered as an indicator of 3-NP neurotoxicity due to depressed energy metabolism. 38 Supporting the above observation, over the course of our study, 3-NP–treated animals showed decreases in body weight with significantly lower feed intakes, which was significantly improved by 14 days pretreatment with lutein, suggesting its therapeutic potential.
Neurobehavioral studies are considered as the most important initial tests for diagnosing chorea and cognitive impairment, which are major symptoms of HD. The main function of basal ganglia is to control the overall coordination of body movements. As the striatum is the central core area in the basal ganglia that controls coordination of motor movement, it is accepted that the 3-NP induces striatal degeneration leading to impaired coordination of motor movement. 39,40 In the present study, 3-NP–induced motor dysfunction was assessed by locomotor and hind-limb impairment, which was significantly improved by lutein pretreatment compared to the 3-NP–treated group.
The memory of HD patients often declines with degeneration of neurons in the brain. Cognitive impairment was suggested to reflect a combination of lower energy levels resulting from SDH inhibition by 3-NP and consequent short term changes in neural processing. In the present sets of experiments, memory performance was tested through the Morris water maze and Elevated Plus maze paradigms. Lutein treatment significantly improved cognitive task performance in both tests, suggesting the potential effect of lutein against 3-NP–induced memory dysfunction.
Apart from neurobehavioral studies, several biochemical parameters, including mitochondrial respiratory chain enzymes and antioxidant parameters, were estimated as a predictor for pathological changes in HD. Oxidative stress and mitochondrial dysfunction induced by 3-NP is mainly responsible for HD-like symptoms seen in rats (chorea and cognitive impairment). 5 Oxidative stress is one of the major deleterious events which contribute to the pathogenesis of several neurodegenerative diseases, including HD. 33,41 3-NP binds irreversibly to complex II (SDH) and leads to inhibition of free fatty acid oxidation pathways; inhibition of these pathways leads to the energy impairment that follows uncoupling of oxidative phosphorylation. Disruption of mitochondrial activity is associated with the abnormal formation of ROS. Inhibition of enzymes in the ETC can lead to an increases in electron leakage from the mitochondria and production of ROS such as the superoxide radical (O2 −•), H2O2, reactive nitrogen species (NO−) and the hydroxyl radical (OH−•), and consequently causes oxidative stress. 28 According to previous reports 42,43 and the clear suggestion of the data in the present study, 3-NP administration significantly increased oxidative stress, as evident by increases in ROS production, nitrite levels, and elevated lipid peroxidation products, and depletion of reduced GSH in 3-NP–treated rats. Oral administration of lutein to 3-NP–treated animals for 14 days significantly attenuated the ROS generation by reversing these parameters in a dose-dependent manner. Catalase is an enzyme which is an endogenous defense against ROS generation. The activity of this enzyme was found to be lowered in 3-NP–treated animals compared with the vehicle-treated group. In contrast, pretreating 3-NP–challenged rats with lutein for 14 days significantly increased levels of catalase. Per se treatment of lutein also increased catalase levels as compared to control treatment, demonstrating that the capability of lutein to increase antioxidant enzyme (catalase) levels might be responsible to a greater extent for its protective role against ROS.
Cognitive impairment is one of the major symptoms of HD. Reduced activity of acetylcholine and choline acetyltransferase has been observed in the brain of HD patients, and is responsible for cognitive deficits. 44 It is reported that 3-NP treatment of rats results in increased activity of the AChE enzyme, which decreases the level of acetylcholine. 45 In the present study, lutein treatment ameliorated the 3-NP–induced increases in AChE activity, which is mainly responsible for cognitive deficits. In addition, AchE-inhibitory activity of lutein is also supported by a clinical study. 12 All of these reports suggest that the AchE-inhibitory property of lutein might be responsible for cognitive improvement offered by lutein as seen in neurobehavioral studies.
Mitochondria of HD patients are affected by alterations in ETC function, in which mitochondrial respiratory chain enzymes (complexes I, II, and IV) are affected. 41 Complex I (NADH dehydrogenase) is the entry enzyme of the mitochondrial respiratory chain and as such plays a crucial role in ATP production and mitochondrial function in general. Complex I impairment leads to increased ROS production in sub mitochondrial particles and cell systems, and is the main site for superoxide formation, while complex IV (cytochrome oxidase) is the last enzyme in the respiratory electon transport chain and responsible for hydroxyl radical formation. In our study, the activity of complex II (SDH dehydrogenase) was decreased to a greater extent as compared to other complexes, and this might be because of irreversible inhibition of complex II by 3-NP. It is the only enzyme that participates in both the citric acid cycle and the ETC; thus, inhibition of this enzyme is responsible for effectively decreasing production of ATP. Based on the assumption that mitochondrial SDH contributes to MTT reduction, the decreased MTT reduction confirms inhibition of SDH. 46 MTT reduction takes place only when reductase enzymes are active, and hence, conversion was used to measure viable cells. This process of conversion mainly affects the timing of when oxidative damage impairs metabolic function, including the ability of mitochondria to make ATP. 47
According to several reports, 3-NP depleted several respiratory chain enzymes (I, II, and IV) and declined the number of viable cells (MTT reduction). 3-NP disturbed mitochondrial enzyme functions, induced damage to the cell-signaling pathway in the brain, and impaired ATP in mitochondria. 34,48 In the present study, these alterations in mitochondrial enzyme complex activities and viable cell counts induced by 3-NP were significantly restored by lutein pretreatment. However, per se treatment of lutein attenuated complex I (NADH dehydrogenase) activity significantly compared with vehicle treatment, but activities of other enzymes (II, IV, and MTT reduction) were not altered significantly, showing lutein's greater propensity towards complex I enzymes than others. In conclusion, lutein restores mitochondrial complex activity, thus preserving normal ATP function and halting cell death induced by 3-NP.
To further investigate the neuroprotective action of lutein on 3-NP–induced pathological lesions, histopathological examinations were performed. As per the reports of histopathological examination, 3-NP treatment produced severe degeneration of neurons, with complete loss of cell detail and architecture and with the appearance of basophilic nuclear remnants in the central zone. Lutein pretreatment (50 mg/kg) showed mild focal gliosis accompanied by mild neuronal degeneration, whereas (100 mg/kg) treatment showed only mild focal gliosis. Thus, the histopathological findings have reconfirmed the protective action of lutein. Brain striatum of the lutein per se treated group showed normal histopathological structure. This indicates that lutein does not possess any adverse effects under normal conditions.
In summary, the neurobehavioral, biochemical and histopathological investigations demonstrate that lutein is able to ameliorate 3-NP induced neurotoxicity by its capability to protect against behavioral changes, restores antioxidant defense enzyme in brain and improves mitochondrial enzymes levels.
Conclusion
Results of the present study demonstrate that chronic treatment with lutein can attenuate HD-like symptoms in animals. Lutein treatment mitigates behavioral, biochemical and histological abnormalities caused by mitochondrial toxin 3-NP in rats by attenuating free radical damage, and therefore could be employed in the management of HD. Future research should therefore be directed to investigate whether our findings using an experimental animal model of HD would have clinical implications in management of HD in human patients.
Footnotes
Acknowledgment
The authors gratefully acknowledge the financial support of AICTE, New Delhi, India.
Author Disclosure Statement
No competing financial interests exist.