
Abstract
Protocatechualdehyde (PCA) is a natural polyphenol compound isolated from the root of the herb S. miltiorrhiza and barley tea plants. PCA possesses antiproliferative and pro-apoptotic properties in human colorectal cancer cells. However, the cellular mechanism has not been fully understood. β-catenin and cyclin D1 are proto-oncogene that is overexpressed in many types of cancers and leads to cancer development. The present study was performed to elucidate the molecular mechanism by which PCA stimulates cell growth arrest and apoptosis in human breast cancer cells. PCA repressed cell proliferation and induced apoptosis in dose-dependent manner. PCA suppressed the expression of β-catenin and cyclin D1 with no changes in mRNA levels. Inhibition of proteosomal degradation using MG-132 and Ada-(Ahx)3-(Leu)3-vinyl sulfone ameliorates PCA-induced downregulation of β-catenin and cyclin D1. PCA treatment decreased the half-life of β-catenin and cyclin D1. PCA-mediated β-catenin downregulation depends on GSK3β. We further provide the evidence that PCA increased nuclear translocation of nuclear factor kappa-B (NF-κB) and the blockage of NF-κB using Bay11-7082 inhibited PCA-mediated β-catenin downregulation. The current study demonstrates that PCA suppress β-catenin expression through GSK3β- and NF-κB-mediated proteosomal degradation. In addition, PCA decreased cyclin D1 expression independent to β-catenin through proteosomal degradation.
Introduction
I
Protocatechualdehyde (PCA) is a (3,4-dihydroxy-benzaldehyde)-polyphenolic compound derived from the root of the herb S. miltiorrhiza 2 and barley tea. 3 PCA has been reported to possess antioxidant, antitumor, and anti-inflammatory properties. 4,5 In human colorectal cancer, PCA suppressed cell proliferation and downregulated cyclin D1 and histone deacetylase. 2,6
Many signaling pathways controls breast tumorigenesis. Specifically, the Wnt and PI3K signaling pathways play important roles in tumorigenesis and cancer progression. 7,8 The β-catenin protein interacts with several cellular proteins including glycogen synthase kinase (GSK) 3β, adenomatous polyposis coli (APC), casein kinase 1α (CK1α), or axin. 9 In particular, GSK3β is inactivated by the PI3K/Akt-mediated phosphorylation at serine 9 residue, and leads to β-catenin accumulation in the cytosol. 10 Free β-catenin translocate into nucleus where β-catenin protein binds to the members of the LEF/TCF family and activates the expression of several target genes, including cyclin D1. 11,12 Thus, activation of β-catenin signaling leads to the induction of cyclin D1 at the transcriptional level and furthers the progression of the G1 to S phase and the increase of proliferation.
In this study, we propose anticancer activity of PCA in breast cancer. PCA inhibits cell growth and increases apoptosis of breast cancer cells by downregulating β-catenin and cyclin D1 via multiple mechanisms.
Materials and Methods
Chemicals
PCA was purchased from Sigma Aldrich (St. Louis, MO, USA), and cell culture media (Dulbecco's modified Eagle medium [DMEM]/F-12) was purchased from Lonza (Walkersville, MD, USA). Antibodies for β-catenin (#9562) and PARP (#9542), phospho GSK3β (Ser9, #9336), GSK-3β (#9315), p65 (#8242), TBP (#8515), and β-actin (#5125) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody for cyclin-D1 (sc-718) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Ada-(Ahx)3-(Leu)3-vinyl sulfone and BAY 11-7082 were purchased from Enzo Life Sciences (Farmingdale, NY, USA). MG-132 and cyclohexamide were purchased from Calbiochem (San Diego, CA, USA). Unless otherwise designated, all chemicals were purchased from Fisher Scientific (Hampton, NH, USA).
Cell culture and treatment
Human breast cancer cell (MCF-7 and MDA-MB-231) was purchased from American Type Culture Collection (Manassas, VA, USA) and was grown in DMEM/F-12 supplemented with 10% fetal bovine serum (FBS) and a mixture of antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin). The cells were maintained in a 37°C incubator at 5% CO2. PCA was dissolved in dimethyl sulfoxide (DMSO) and treated to the cells. DMSO was used for a vehicle and the final concentration of DMSO did not exceed 0.1% (v/v). The cells were pre-treated with various inhibitors (MG-132, Ada-(Ahx)3-(Leu)3-vinyl sulfone, SB216763, Bay11-7082) and then co-treated with 100 μM of PCA for 24 hours as indicated in legends of Figures 3B, 3C, 4A and 5B. For cycloheximide experiment, the cells were pre-treated with DMSO or PCA for 6 hours and then co-treated with 10 μg/mL of cycloheximide for different times as indicated in legends of Figures 3D and 3E.
Measurement of cell proliferation
Cell Proliferation Assay system (Promega, Madison, WI, USA) was used for measuring proliferative activity of the cells. MCF-7 cells (2000 cells/well) were plated onto 96-well plates. On the next day, the cells were treated with 0, 5, 10, 25, 50, or 100 μM of PCA in media containing 10% FBS for 0, 24, or 48 hours. Twenty microliters of CellTiter96 Aqueous One solution was added to each well and further incubated for 1 hour at 37°C. The absorbance was measured at 490 nm using enzyme-linked immunosorbent assay plated reader (Bio-Tek Instrument Inc., Winooski, VT, USA).
Measurement of apoptosis
Flow cytometric detection of apoptotic cells was performed according to the manufacturer's instructions of Annexin V kits (Trevigen, Gaithersburg, MD, USA). Briefly, the cells were plated in 6-well tissue culture dishes and treated with 0, 50, and 100 μM of PCA for 24 hours. The attached and floating cells were harvested together with trypsin-EDTA. After washing with phosphate-buffered saline (PBS), the whole cells were resuspended and collected by centrifugation at 1000 rpm for 5 minutes. Cell pellets were stained with Annexin V-FITC and early apoptotic cells were quantified in FACS co-facility at University of Maryland using FACSCanto II (Becton Dickinson Biosciences, San Jose, CA, USA).
Isolation of cytosol and nucleus fraction
Cytosol and nucleus fractions were extracted from cells according to manufacturer's protocols of a nuclear extract kit (Active Motif, Carlsbad, CA, USA). Briefly, the cells were harvested with 1X hypotonic buffer and incubated for 15 minutes on ice. After centrifugation for 30 seconds at 14,000 g, the supernatants (cytoplasmic fraction) were isolated. The nuclear pellet was incubated with lysis buffer for 30 minutes at 4°C under agitation. The nuclear suspension was centrifuged for 10 minutes at 14,000 g and the supernatant (nuclear fraction) was collected. Both nuclear and cytoplasmic fractions were used for Western blot analysis.
Isolation of RNA and reverse-transcription polymerase chain reaction
Total RNA was extracted by RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to manufacturer's protocol. Briefly, the cells were lysed in RNeasy lysis buffer containing β-mercaptoethanol and then added 70% ethanol to the homogenized lysate. The total RNA bound to membrane of RNeasy spin column was eluted in RNase-free water. The 1 μg of total RNA was used for reverse-transcription (RT) reaction according to manufacturer's protocol (Verso cDNA kit, Thermo Scientific, Pittsburg, PA, USA). Polymerase chain reaction (PCR) was conducted as follows: total 30 cycles at 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute using PCR Master Mix kit (Promega). The primer sequences are as follows: β-catenin (CTNNB1): forward 5′-CCC ACT AAT GTC CAG CGT TT-3′ and reverse 5′-AAT CCA CTG GTG AAC CAA GC-3′; cyclin D1: forward 5′-AAC TAC CTG GAC CGC TTC CT-3′ and reverse 5′-CCA CTT GAG CTT GTT CAC CA-3′; GAPDH: forward 5′-ACC CAG AAG ACT GTG GAT GG-3′ and reverse 5′-TTC TAG ACG GCA GGT CAG GT-3′.
SDS-PAGE and Western blot
To prepare samples for electrophoresis, cells were lysed in radio immuno-precipitation assay (RIPA) buffer (Boston Bioproduct Inc., Ashland, MA, USA) with mixture of protease inhibitors and phosphatase inhibitors (Sigma Aldrich, St. Louis, MO, USA). The protein concentration of cell lysates was determined by the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL, USA) using a bovine serum albumin (BSA) as a standard. The samples were denatured with 1X loading buffer at 95–100°C for 5 minutes. The proteins were separated on the sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) and transferred to the nitrocellulose transfer membranes (Whatman GmbH, Dassel, Germany). The membrane was blocked using 5% non-fat milk to prevent non-specific background and incubated with the primary antibodies at 4°C overnight. The next day, the membrane was incubated with the secondary antibody conjugated with horseradish peroxidase (HRP) for 1 hour at room temperature. Using Pierce ECL Western blotting substrate (Thermo Scientific, Rockford, IL, USA), chemiluminescence was detected and imaged by ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Statistical analysis was measured with the Student's unpaired t-test, with statistical significance set at P<.05.
Results
PCA inhibits proliferation and stimulates apoptosis of MCF-7 cells
To investigate whether or not the treatment of PCA affects the growth of breast cancer cells, we tested estrogen receptor (ER)-positive (MCF-7) and negative (MDA-MB-231) breast cancer cells. Both cells were exposed to 0, 5, 10, 25, 50, and 100 μM of PCA for 0, 24, and 48 hours into media containing 10% serum. Thereafter, cell proliferation was measured. As shown in Figure 1A (top), MCF-7 cells treated with 50 μM and 100 μM of PCA significantly decreased cell growth by 11% and 20% in 24 hours and by 22% and 27% in 48 hours, respectively, when compared to the cells treated with 0 μM of PCA. However, PCA did not change the cell proliferation in MDA-MB-231 cells (Fig. 1A, bottom). Next, we explored whether PCA affect apoptosis of MCF-7 cells. The cells were exposed to 0, 50, and 100 μM of PCA for 24 hours and early apoptotic cells were detected with FACS analysis (Fig. 1B). As a result, the percentage of early apoptotic cells were increased by 1.9-fold and 2.6-fold in the cells treated with 50 μM and 100 μM of PCA, respectively, when compared to the cells treated with 0 μM of PCA. These results indicate that PCA may inhibit cell proliferation and increases apoptosis in human breast cancer cells through ER-dependent manner because PCA suppresses proliferation of estrogen receptor (ER)-positive (MCF-7) breast cancer cells, but not ER-negative (MDA-MB-231) breast cancer cells.
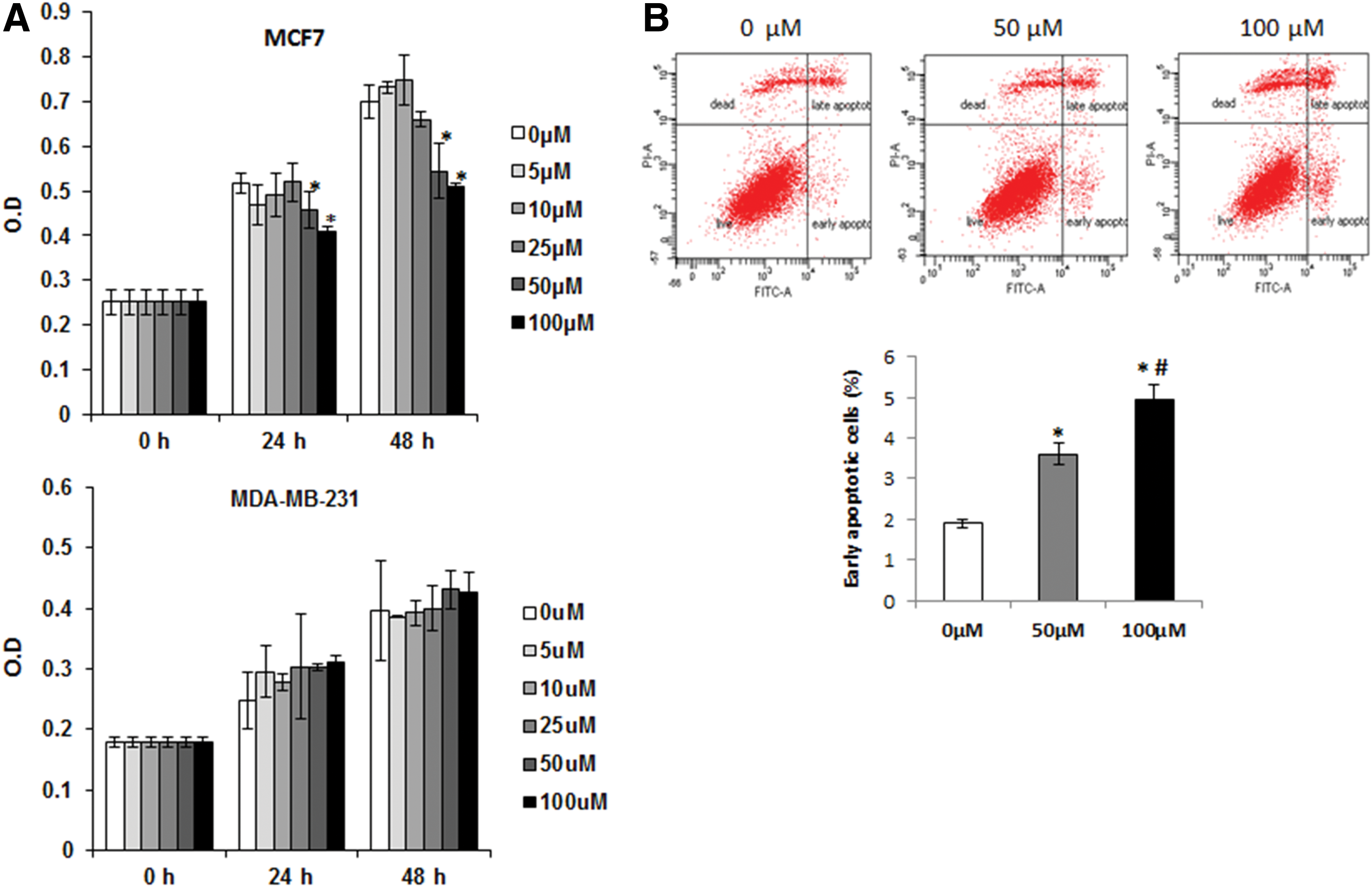
Inhibition of cell proliferation and induction of apoptosis by protocatechualdehyde (PCA).
Inhibitory effect of PCA on β-catenin and cyclin D1 expression
Next, we measured PARP cleavage, a marker of apoptosis. As shown in Figure 2A and 2B, the amount of cleaved fractions increased in the cells treated with PCA in dose- and time-dependent manners. It is generally accepted that an induction of apoptosis is associated with Wnt signaling pathway. Thus, in order to test whether PCA treatment affects β-catenin protein level, Western blot was performed. As shown in Figure 2A and B, treatment of 50 μM and 100 μM PCA to MCF-7 cells resulted in a decrease of β-catenin protein level in a dose- and a time-dependent manner. Increased β-catenin translocates to the nucleus, and binds TCF/LEF transcription factor and regulates transcription of downstream target gene such as cyclin D1. 12 Therefore, we reblotted the same membrane with antibody for cyclin D1. The protein level of cyclin D1 is also decreased at the cells treated with 50 μM and 100 μM of PCA and decreased 24 hours after treatment of 100 μM of PCA. These data propose the potential that antiproliferative and pro-apoptotic activity of PCA might be mediated via decreased expression of β-catenin and cyclin D1.
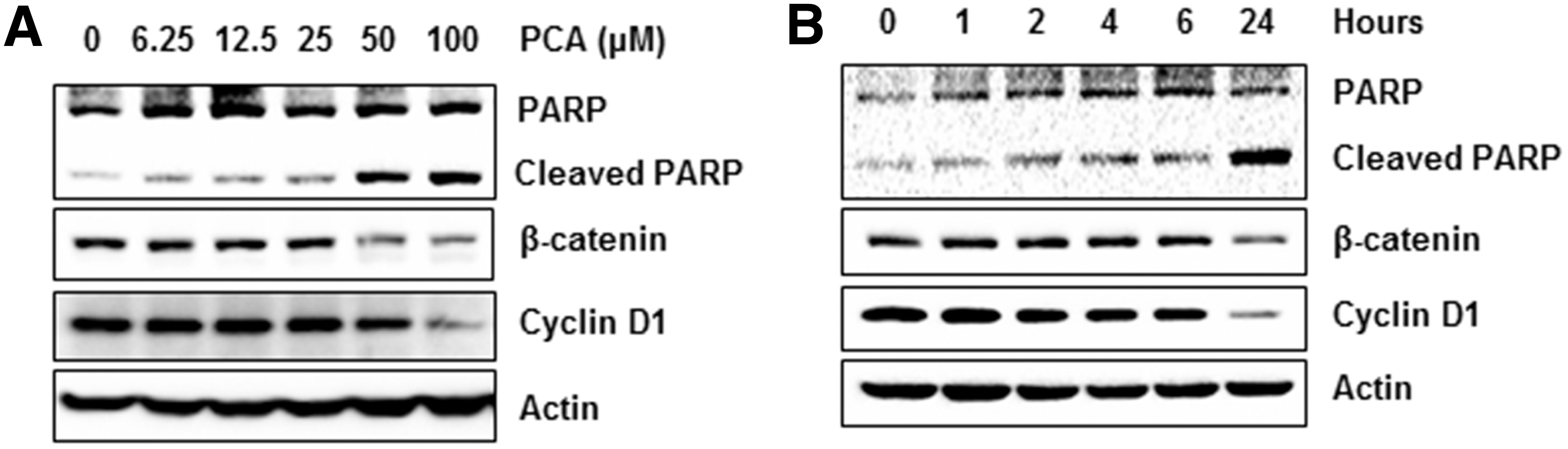
Downregulation of β-catenin and cyclin D1 by PCA.
Proteosomal degradation of β-catenin and cyclin D1 by PCA
To investigate whether or not an inhibition of β-catenin protein level may be associated with transcriptional downregulation of β-catenin gene, MCF-7 cells were incubated with 0, 50, and 100 μM of PCA for 24 hours and RT-PCR was performed to measure β-catenin mRNA. As shown in Figure 3A, mRNA level was not different among treatments. Interestingly, cyclin D1 mRNA was not affected by PCA treatment, indicating that PCA may decrease protein level of β-catenin and cyclin D1 through enhancing proteosomal degradation.
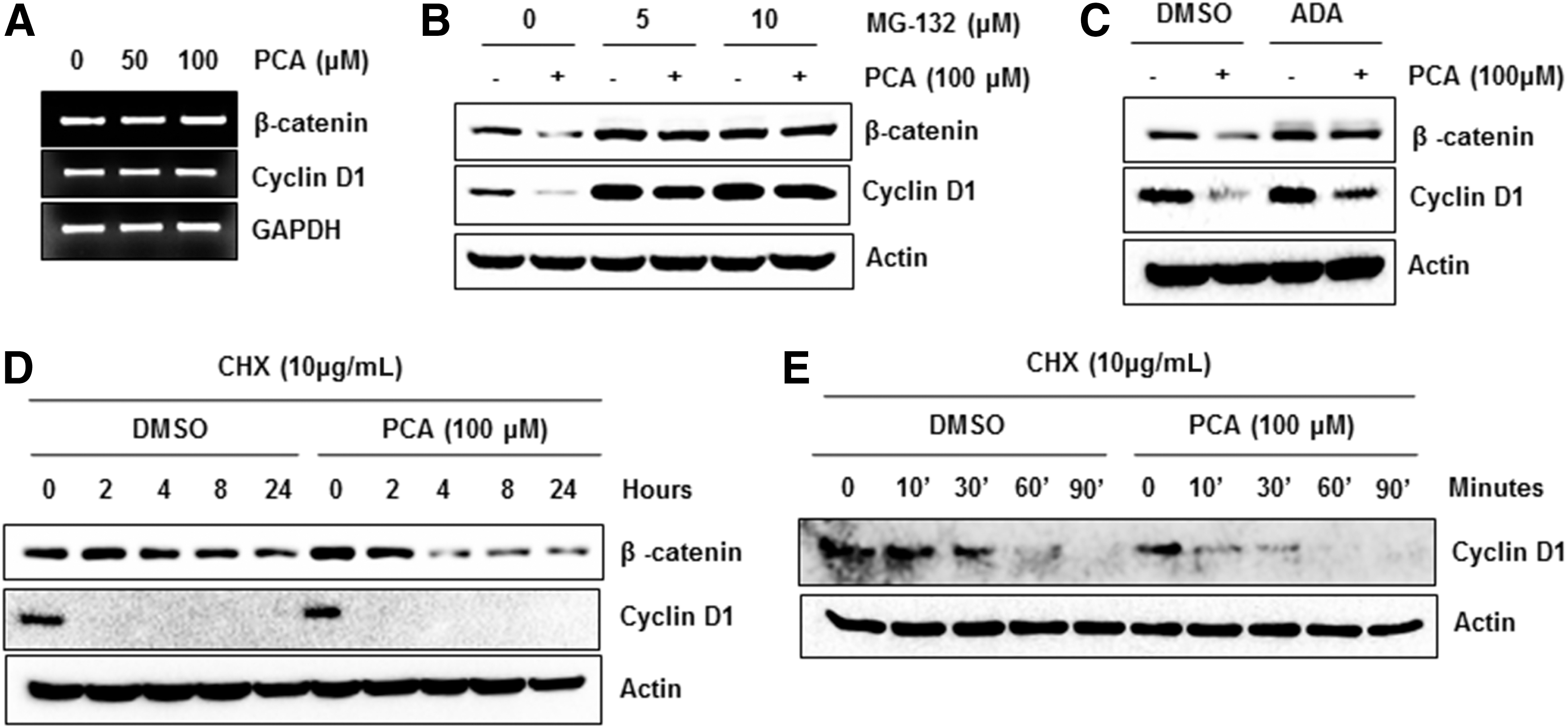
Proteosomal degradation of β-catenin and cyclin D1 by PCA.
Thus, we performed experiments using the proteasome inhibitor, MG-132 and Ada-(Ahx)3-(Leu)3-vinyl sulfone (ADA). As shown in Figure 3B and 3C, pre-treatment of MG-132 and ADA blocked PCA-stimulated downregulation of β-catenin and cyclin D1. To confirm this data, we pre-treated the cells with DMSO or PCA and then co-treated with cycloheximide (CHX), a protein synthesis inhibitor, for indicated times. We observed that β-catenin protein was degraded more rapidly in the cells treated with PCA than vehicle-treated control (Fig. 3D). Because half-life of cyclin D1 was very short after CHX treatment, we performed another experiment exposing the cells to CHX for short times. As shown in Figure 3E, PCA treatment decreased cyclin D1 more rapidly than DMSO-treated control. Overall, the data indicate that proteosomal degradation might be responsible mechanism for downregulation of β-catenin and cyclin D1 by PCA.
β-catenin downregulation by PCA is depending on GSK3β
Cellular β-catenin level is regulated by PI3K/AKT/GSK3β pathway. Activation of glycogen synthase kinase3β (GSK3β) induces destabilization of β-catenin and subsequent proteosomal degradation. 10 In order to investigate whether GSK3β mediates PCA-induced downregulation of β-catenin, we pretreated the MCF-7 cells with specific inhibitor for GSK3β (SB216763) and then co-treated with DMSO or PCA. As shown in Figure 4A, pretreatment with SB216763 suppressed PCA-mediated downregulation of β-catenin. Since phosphorylation of GSK3β (p-GSK3β) at residue Ser9 decrease enzymatic activity of GSK3β, we measured phosphorylation of Ser9. As you can see in Figure 4B, inactive GSK3β decreased by treatment with PCA for 24 hours (Fig. 4B) without a change of total GSK3β. These results suggest that GSK3β, as a downstream target of PK3/AKT pathway, might play a pivotal role for PCA-mediated downregulation of β-catenin.
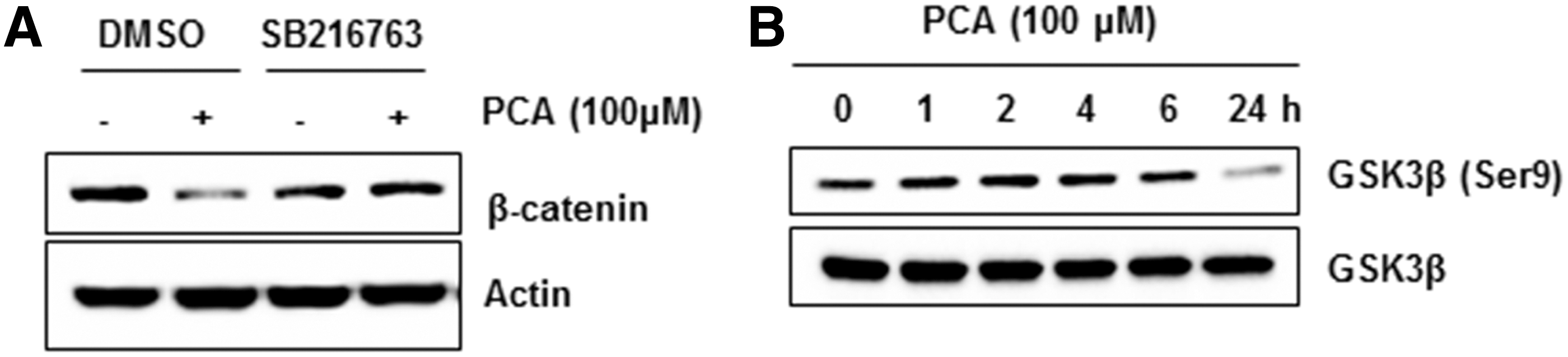
GSK3β dependency of β-catenin downregulation by PCA.
PCA induces activation of nuclear factor kappa-B pathway
There is evidence to suggest that the nuclear factor kappa-B (NF-κB) activation is associated with β-catenin and affect development and progression of cancer. To test whether PCA treatment influences NF-κB pathway, we observed the expression of p65 in cytosolic and nuclear fraction of MCF-7 cells after treatment of PCA. As a result, we found that PCA increased an amount of p65 in nucleus of MCF-7 cells (Fig. 5A). According to recent literatures, NF-κB interacts with β-catenin in several proposed mechanisms. 13,14 Thus, to test if NF-κB mediates PCA-stimulated downregulation of β-catenin, MCF-7 cells were pre-treated with BAY 11-7082, selective inhibitor of NF-κB translocation and then co-treated with DMSO or PCA. As shown in Figure 5B, downregulation of β-catenin by PCA was ameliorated by pretreatment of BAY 11-7082, indicating that activation of NF-κB by PCA at least in part contributes PCA-mediated downregulation of β-catenin in human breast cancer cells.
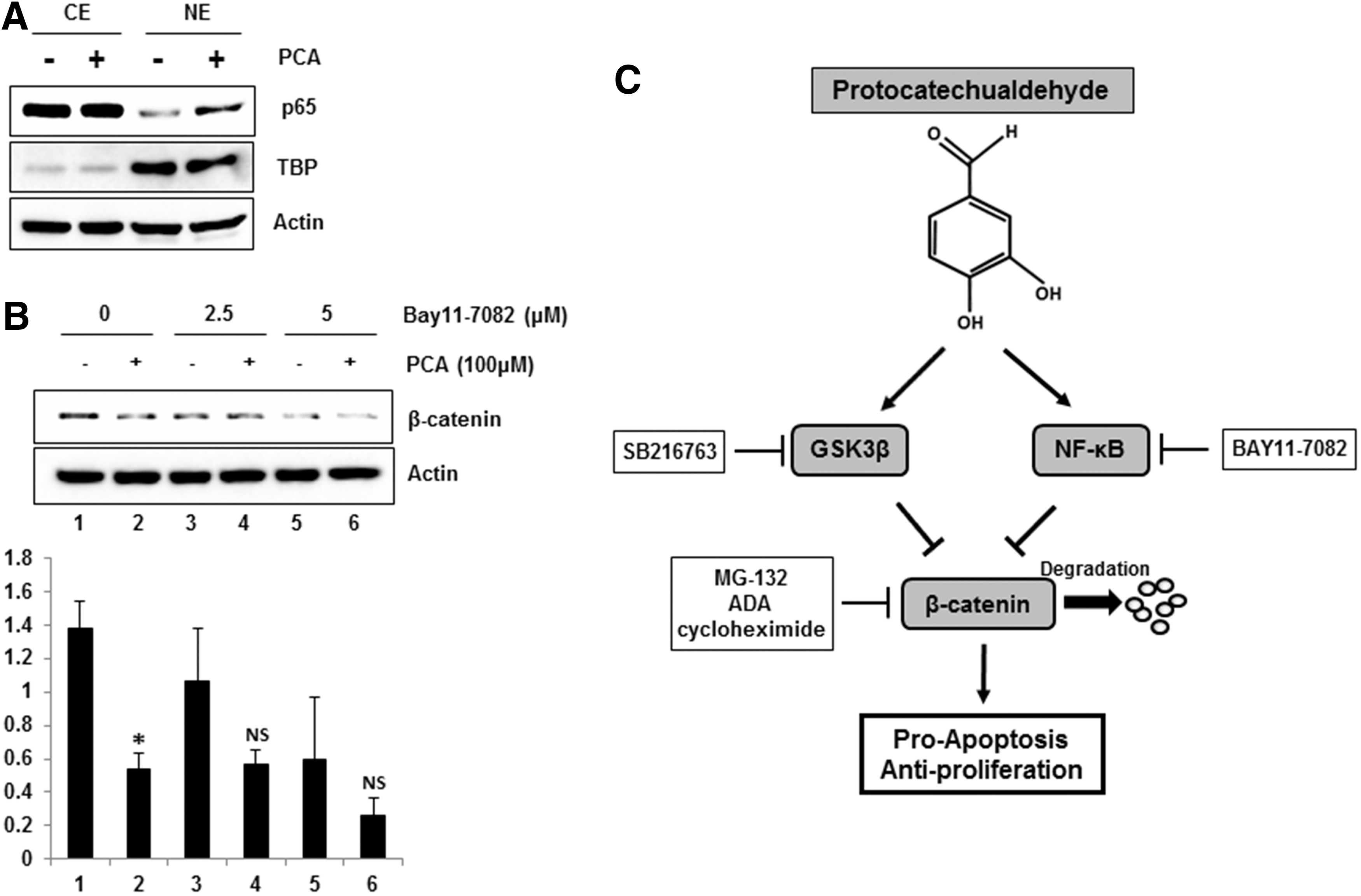
NF-κB dependency of β-catenin downregulation by PCA.
Discussion
PCA is a naturally occurring polyphenol in plants. There were several reports that PCA possesses anticancer activity. 15,16 In previous study, we reported that PCA inhibited proliferation of human colorectal cancer cells. 6 The anti-carcinogenic effects of PCA have raised an important question regarding the underlying molecular mechanisms. Here, we demonstrate direct evidence that PCA downregulates β-catenin and cyclin D1 through activating proteosomal degradation. The proteosomal degradation of β-catenin is associated with GSK3β and NF-κB pathway in human breast cancer cells.
β-catenin is regulated by several pathways and one is that β-catenin revitalize as a transcriptional activator via a Wnt signaling pathway. 17 GSK3 is a downstream target of PI3K/AKT and Wnt signaling pathways and has recently been recognized as one of the most important signals ensuring apoptosis in the cancer cells. 10 Deregulation of PI3K/AKT/GSK3β pathways by specific inhibitor triggers a cellular cascade involved in apoptosis in various cancer model. 18,19 The present data indicate that activation of GSK3β may be a major mechanism by which PCA downregulates β-catenin.
The effect of PCA on β-catenin/TCF-dependent gene regulation can be important for the PCA-induced anti-tumorigenesis. The importance of this effect is clearly indicated by the reduced expression of cyclin D1, a protein that plays an important role in cell-cycle transitions. In fact, cyclin D1 overexpression has been found in approximately 50% of breast cancer. 20 Our data indicate that PCA-induced cyclin D1 downregulation is independent on β-catenin downregulation and completely related to its proteosomal degradation. There are multiple mechanisms by which dietary compounds represses cyclin D1 expression. One is through transcriptional regulation. In the previous study, we found that PCA decrease cyclin D1 in transcriptional downregulation. 6 In fact, TCF/LEF site is located in this region of the promoter and plays an important role in β-catenin-dependent transcriptional regulation of cyclin D1. 12 Another mechanism to suppress cyclin D1 expression is the activation of proteasome degradation. Our results clearly demonstrate that PCA increase proteosomal degradation. Proteolysis of cyclin D1 is one of prominent anticancer mechanism, previously reported with curcumin, 21 retinoic acid, 22 and troglitazone. 23
NF-κB has been known as a potential molecular target for inflammation and cancer. 24,25 Transcriptional activity of NF-κB is determined by cellular localization of two nuclear transcription factors, p50 and p65. Without stimulus, p65 and p50 is sequestered in IκB-α, but activation of NF-κB signaling induce proteosomal degradation of IκB-α by activating IκB-α kinase (IKK), and increase translocation of p65 and p50 to the nucleus where coordinate transcription of several target genes associated with inflammation, cell proliferation, and apoptosis. 26 In our study, we observed that PCA treatment increase nuclear translocation of p65 (Fig. 5A). The role of NF-κB in cancer development is complex. According to several recent studies, NF-κB activation mediates apoptosis of anticancer compounds. 27 –29 It is proposed that NF-κB activation is a cancer preventive and therapeutic target. In addition, NF-κB interacts with β-catenin and may control cancer progression. β-catenin forms a complex with NF-κB and reduces NF-κB transactivation in human breast cancer. 14 Therefore, it is likely that NF-κB activation by PCA might be at least a partly responsible mechanism for β-catenin downregulation. In fact, NSAIDs such as diclofenac inhibited β-catenin signaling through activating NF-κB. 30
Many studies indicated that estrogen via estrogen receptor (ER)-mediated signaling pathways participate in cancer development and progression by modulating expressions of genes involved in cell cycles and apoptosis. Our data demonstrate that PCA suppressed proliferation of breast cancer cells via estrogen-dependent manner (Fig. 1A). In fact, β-catenin is highly associated with estrogen and inhibition of ER activation could markedly suppress growth of breast cancer cells through modulation of several signaling pathways. 31,32 Thus, it is likely that link of β-catenin and estrogen signaling is crucial for PCA-stimulated suppression of breast cancer cell growth. In conclusion, PCA activates GSK3β by dephosphorylating serine 9 and activates NF-κB pathway by increasing nuclear translocation of p65. Activation of GSK3β and NF-κB subsequently stimulate proteosomal degradation of β-catenin and inhibit proliferation and induces apoptosis in human breast cancer cells (Fig. 5C). The current study provides information on molecular events of anticancer activity of PCA.
Footnotes
Acknowledgments
This work was supported by start-up funds (S.-H.L.) from University of Maryland and in part by #RSG-11-133-01-CCE (S.-H. L.) from the American Cancer Society.
Author Disclosure Statement
There are no competing financial interests.