
Abstract
The excessive release of glutamate is a critical element in the neuropathology of acute and chronic brain disorders. The purpose of the present study was to investigate the effect and possible mechanism of myricetin, a naturally occurring flavonoid with a neuroprotective profile, on endogenous glutamate release in the nerve terminals (synaptosomes) of the rat cerebral cortex. The release of glutamate was evoked by the K+ channel blocker 4-aminopyridine (4-AP) and measured by one-line enzyme-coupled fluorometric assay. We also used a membrane potential-sensitive dye to assay the synaptosomal plasma membrane potential, and a Ca2+ indicator Fura-2 to monitor cytosolic Ca2+ concentrations ([Ca2+]C). Results show that myricetin inhibited 4-AP-evoked glutamate release, and this effect was prevented by chelating extracellular Ca2+ ions and the vesicular transporter inhibitor bafilomycin A1. However, the glutamate transporter inhibitor
Introduction
F
In the brain, glutamate is a major excitatory neurotransmitter that plays a vital role in numerous brain functions such as synaptic plasticity, learning, and memory. 17 However, in addition to the physiological role of glutamate, excessive glutamate release and activation of the glutamate receptors increase intracellular Ca2+ levels, subsequently triggering a cascade of cellular responses, including enhanced oxygen free radical production, disturbed mitochondrial function, and protease activation, which ultimately kill the neurons. This process has been implicated as a pathophysiological factor in multiple neurological disorders, both acute, such as stroke and head trauma, and chronic, such as neurodegenerative disorders. 18 –20 Thus, inhibiting glutamate release may provide a potential target for neuroprotective action.
Because myricetin has a neuroprotective profile and the excessive release of glutamate is a critical element in the neuropathology of acute and chronic brain disorders, assessing the effects of myricetin on glutamate release is warranted. The purpose of the present study was to use isolated nerve terminals (synaptosomes) from the rat cerebral cortex to determine whether myricetin affects glutamate release. The isolated presynaptic terminal is a well-established model for studying the molecular mechanisms underlying the presynaptic phenomena. Specifically, this preparation is capable of accumulating, storing, and releasing neurotransmitters and does not suffer from any postsynaptic interactions. 21 Using this model, we examined the effects of myricetin on the levels of released glutamate, the synaptosomal plasma membrane potential, and the activation of the voltage-dependent Ca2+ channels (VDCCs).
Materials and Methods
Materials
3′,3′,3′-dipropylthiadicarbocyanine iodide [DiSC3(5)] and fura-2-acetoxymethyl ester (Fura-2-AM) were obtained from Invitrogen (Carlsbad, CA, USA). Bafilomycin A1, ω-conotoxin MVIIC (ω-CgTX MVIIC), dantrolene, 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP37157), and
Animals
Adult male Sprague-Dawley rats (150–200 g) were used in this study. All of the animal experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and approved by the Fu Jen Institutional Animal Care and Utilization Committee. All efforts were made to minimize the number of animals and their suffering.
Synaptosomal preparation
Percoll-purified synaptosomes were prepared using the cerebral cortex of rats, as described previously. 21 –23 Briefly, the rats were sacrificed by decapitation and the brains were removed at 4°C. The cerebral cortex was rapidly dissected and homogenized in a medium containing 0.32 mM sucrose, pH 7.4. The homogenate was centrifuged at 3000 g for 10 min to remove nuclei and debris, and the supernatant was gently stratified on a discontinuous Percoll gradient (3%, 10%, and 23% in sucrose) and centrifuged at 32,500 g for 7 min. The layer between 10% and 20% Percoll (synaptosomal fraction) was subsequently collected and washed by centrifugation. The final synaptosomal pellet was resuspended in 2 mL HEPES-buffered incubation medium (HBM [mM]: NaCl, 140; KCl, 5; NaHCO3, 5; MgCl2·6H2O, 1; Na2HPO4, 1.2; glucose, 10; HEPES, 20; pH 7.4), and the protein concentration was determined using the Bradford assay. 24 Synaptosomes were centrifuged in the final wash to obtain synaptosomal pellets with 0.5 mg of protein. The synaptosomal pellets were stored on ice and used within 2–3 h.
Glutamate release
The glutamate release was assayed by using on-line fluorimetry, as described previously. 25 Synaptosomal pellets (0.3 mg of protein) were resuspended in the HBM containing 16 μM bovine serum albumin (BSA) and incubated in a stirred and thermostated cuvette at 37°C in a Perkin-Elmer LS-55 spectrofluorometer (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA). NADP+ (2 mM), glutamate dehydrogenase (GDH, 50 U/mL), and CaCl2 (1.2 mM) were added after 3 min. After an additional 10 min of incubation, 4-aminopyridine (4-AP, 1 mM) or high external KCl (15 mM) was added to stimulate glutamate release. Glutamate release was monitored by measuring the increase in fluorescence (excitation and emission wavelengths of 340 and 460 nm, respectively), caused by NADPH being produced by the oxidative deamination of released glutamate by GDH. Data were accumulated at 2-s intervals. A standard of exogenous glutamate (5 nmol) was added at the end of each experiment, and the fluorescence response used to calculate the released glutamate was expressed as nmol glutamate/mg synaptosomal protein (nmol/mg). Values quoted in the text and depicted in bar graphs represent the levels of glutamate cumulatively released after 5 min of depolarization and are expressed as nmol/mg/5 min. Cumulative data were analyzed using Lotus 1-2-3.
Synaptosomal plasma membrane potential
The plasma membrane potential was determined using a membrane potential-sensitive dye, DiSC3(5). 26 Synaptosomes were resuspended in the HBM and incubated in a stirred and thermostated cuvette at 37°C in a Perkin-Elmer LS-55 spectrofluorimeter. After 3 min of incubation, 5 μM of DiSC3(5) was added and allowed to equilibrate before the addition of CaCl2 (1.2 mM) after 4 min of incubation. 4-AP (1 mM) was then added to depolarize the synaptosomes for 10 min, and DiSC3(5) fluorescence was monitored at excitation and emission wavelengths of 646 and 674 nm, respectively. Cumulative data were analyzed using Lotus 1-2-3 and expressed in fluorescence units.
Cytosolic free Ca2+ concentration
The cytosolic Ca2+ concentrations ([Ca2+]C) were measured using the Ca2+ indicator Fura-2. Synaptosomes (0.5 mg/mL) were resuspended in the HBM containing 0.1 mM CaCl2 and loaded with 5 μM Fura-2-AM for 30 min at 37°C. The synaptosomes were washed with HBM by centrifugation, resuspended in 2 mL of HBM containing BSA, and placed in a Perkin-Elmer LS-55 spectrofluorometer at 37°C with stirring in the presence of 1.2 mM CaCl2. The synaptosomes were incubated for 10 min in the presence of myricetin (30 μM) before being depolarized with 4-AP (1 mM). Fura-2-Ca fluorescence was determined at excitation wavelengths of 340 and 380 nm (emission wavelength, 505 nm), and data were accumulated at 2-s intervals. [Ca2+]C (nM) was calculated by using calibration procedures 27 and equations, as described previously. 28 Cumulative data were analyzed using Lotus 1-2-3.
Data analysis
Data are expressed as mean±standard error of the mean. The data reported were analyzed by using the unpaired Student's t-test or by using one-way analysis of variance accompanied by post hoc Tukey comparison tests for multiple comparisons. The analysis was completed using SPSS software (17.0; SPSS, Inc., Chicago, IL, USA). P<.05 was considered to represent a significant difference.
Results
To investigate the effect of myricetin on glutamate release, isolated nerve terminals were depolarized with the K+ channel blocker 4-AP. 4-AP destabilizes the membrane potential and is thought to cause repetitive spontaneous Na+ channel-dependent depolarization that closely approximates in vivo depolarization of the synaptic terminal that leads to the activation of VDCCs and neurotransmitter release. 29 Under control conditions, 4-AP (1 mM) evoked glutamate release of 7.2±0.1 nmol/mg/5 min from synaptosomes incubated in the presence of 1.2 mM CaCl2 (basal, 0.07±0.01 nmol/mg/5 min). Application of myricetin (30 μM) produced an inhibition of 4-AP-evoked glutamate release to 3.1±0.1 nmol/mg/5 min (n=8; P<.01), without altering the basal release of glutamate (0.06±0.02 nmol/mg/5 min; Fig. 1A). The inhibitory effect of myricetin on 4-AP-evoked glutamate release was concentration dependent and produced an IC50 value of∼18 μM, which was derived from a dose–response curve (Fig. 1B).
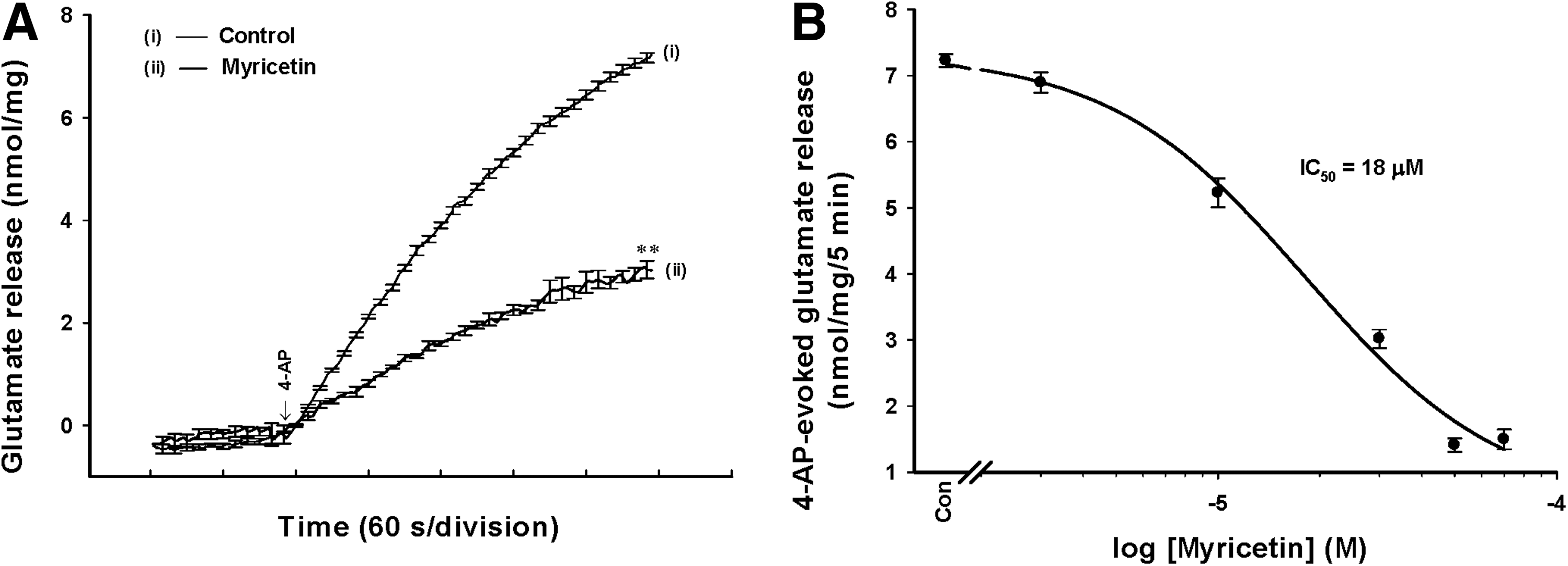
Myricetin inhibits 4-aminopyridine (4-AP)-evoked release of glutamate in rat cerebrocortical nerve terminals.
The 4-AP-evoked release of glutamate from synaptosomes can be sustained by different mechanisms, including exocytosis (Ca2+-dependent release) and reversal of the transporter (Ca2+-independent release).
30,31
To discriminate the effect of myricetin on these two components of endogenous glutamate release evoked by 4-AP, we performed a series of experiments as follows. First, the Ca2+-independent glutamate efflux was measured by depolarizing the synaptosomes with 1 mM 4-AP in the extracellular Ca2+-free solution that contained 300 μM EGTA. Under these conditions, 4-AP (1 mM) induced an increase in glutamate release from 0.04±0.01 nmol/mg/5 min to 1.2±0.2 nmol/mg/5 min (Fig. 2A). The Ca2+-independent release evoked by 4-AP was reduced by 10 μM
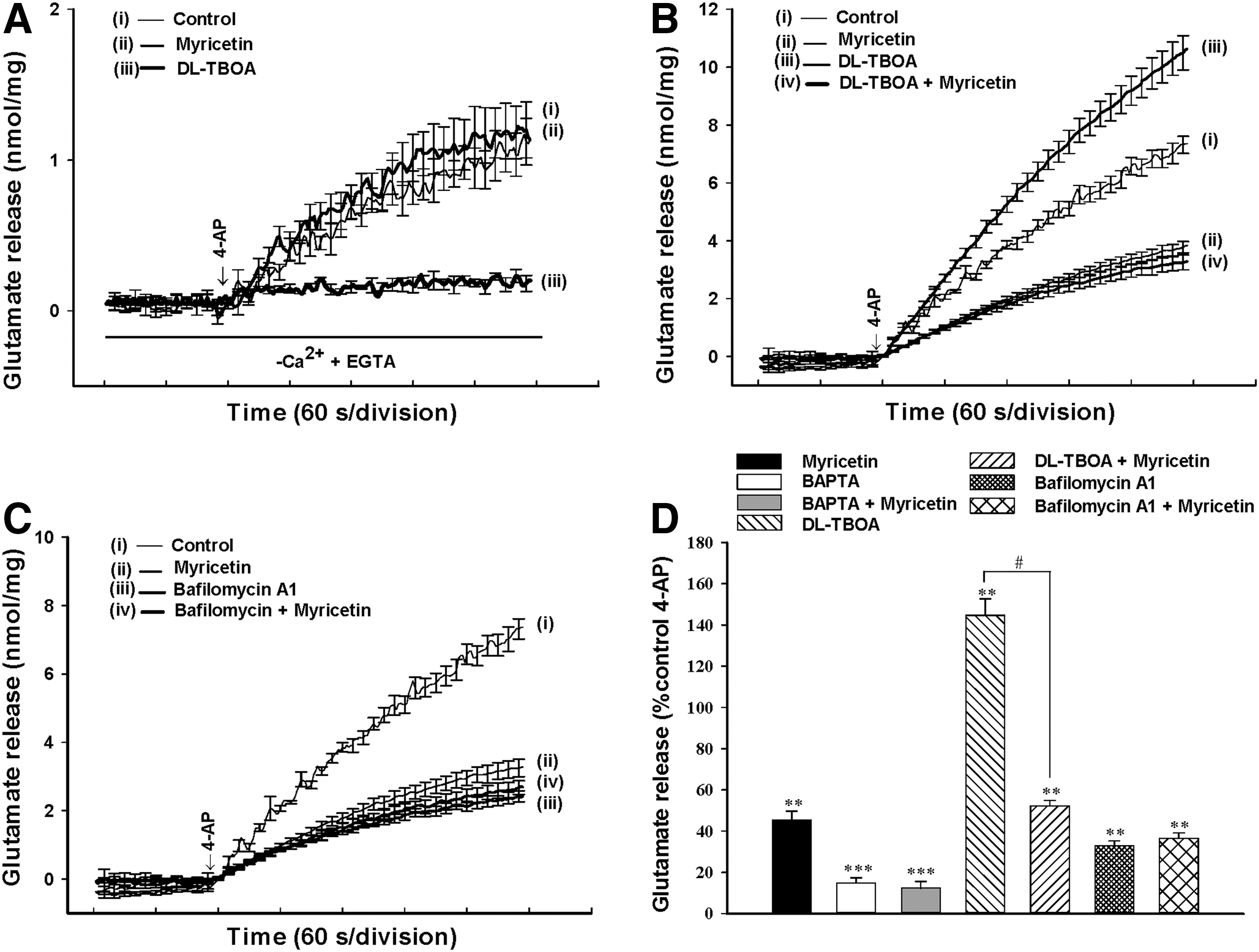
Myricetin effects on the Ca2+-dependent exocytotic component of 4-AP-evoked glutamate release.
To further clarify the mechanism of myricetin-mediated inhibition of glutamate release, we used a membrane potential-sensitive dye, DiSC3(5), to determine the effect of myricetin on the synaptosomal plasma membrane potential. As shown in Figure 3A, 4-AP (1 mM) caused an increase in DiSC3(5) fluorescence of 5.2±0.9 fluorescence units/5 min (n=5). Preincubation of synaptosomes with myricetin (30 μM) for 10 min before 4-AP addition did not alter the resting plasma membrane potential and produced no substantial change in the 4-AP-mediated increase in DiSC3(5) fluorescence (5.1±0.9 fluorescence units/5 min, n=5; P=.963; Fig. 3A). In addition, we confirmed the myricetin-mediated inhibition of glutamate release using an alternative secretagogue, high external [K+]. Elevated extracellular KCl depolarizes the plasma membrane by shifting the K+ equilibrium potential above the threshold potential for activation of VDCCs, which leads to Ca2+ entry and neurotransmitter release, while Na+ channels are inactivated. 32 In Figure 3B, control glutamate release evoked by 15 mM KCl (6.5±0.3 nmol/mg/5 min) was potently inhibited by 30 μM myricetin (2.5±0.2 nmol/mg/5 min; n=5; P<.001).
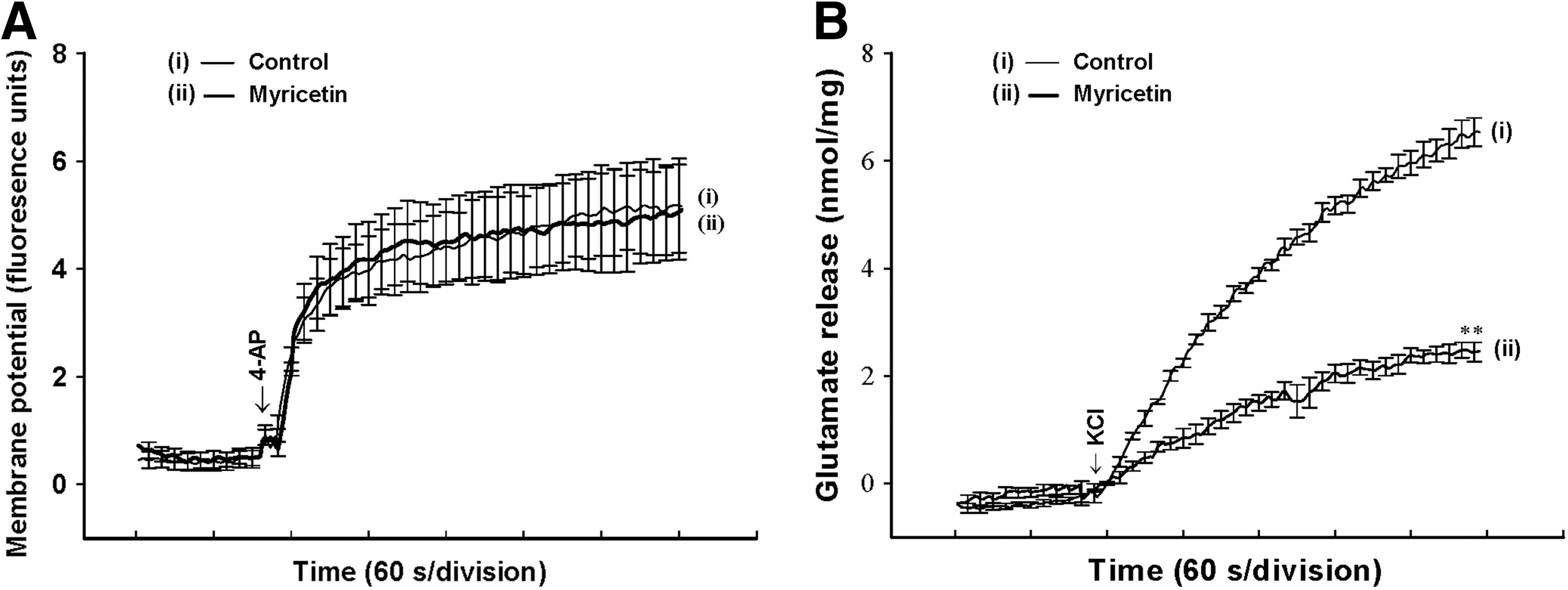
Myricetin does not alter the synaptosomal membrane potential.
Downstream of membrane depolarization, presynaptic inhibition of neurotransmitter release can be mediated by a reduction of [Ca2+]C. To test whether a reduction in [Ca2+]C is responsible for the myricetin-mediated inhibition of release, we determined the [Ca2+]C using the Ca2+ indicator Fura-2. Under control conditions, 4-AP (1 mM) caused a rise in [Ca2+]C, from 118.5±0.2 nM to a plateau level of 199.2±5.7 nM (n=5; Fig. 4). Application of myricetin (30 μM) did not significantly affect basal Ca2+ levels (116.0±0.4 nM), but caused a 40.4±3.2% decrease in the 4-AP-evoked rise in [Ca2+]C (164.1±4.6 nM; P<.01; n=5; Fig. 4).
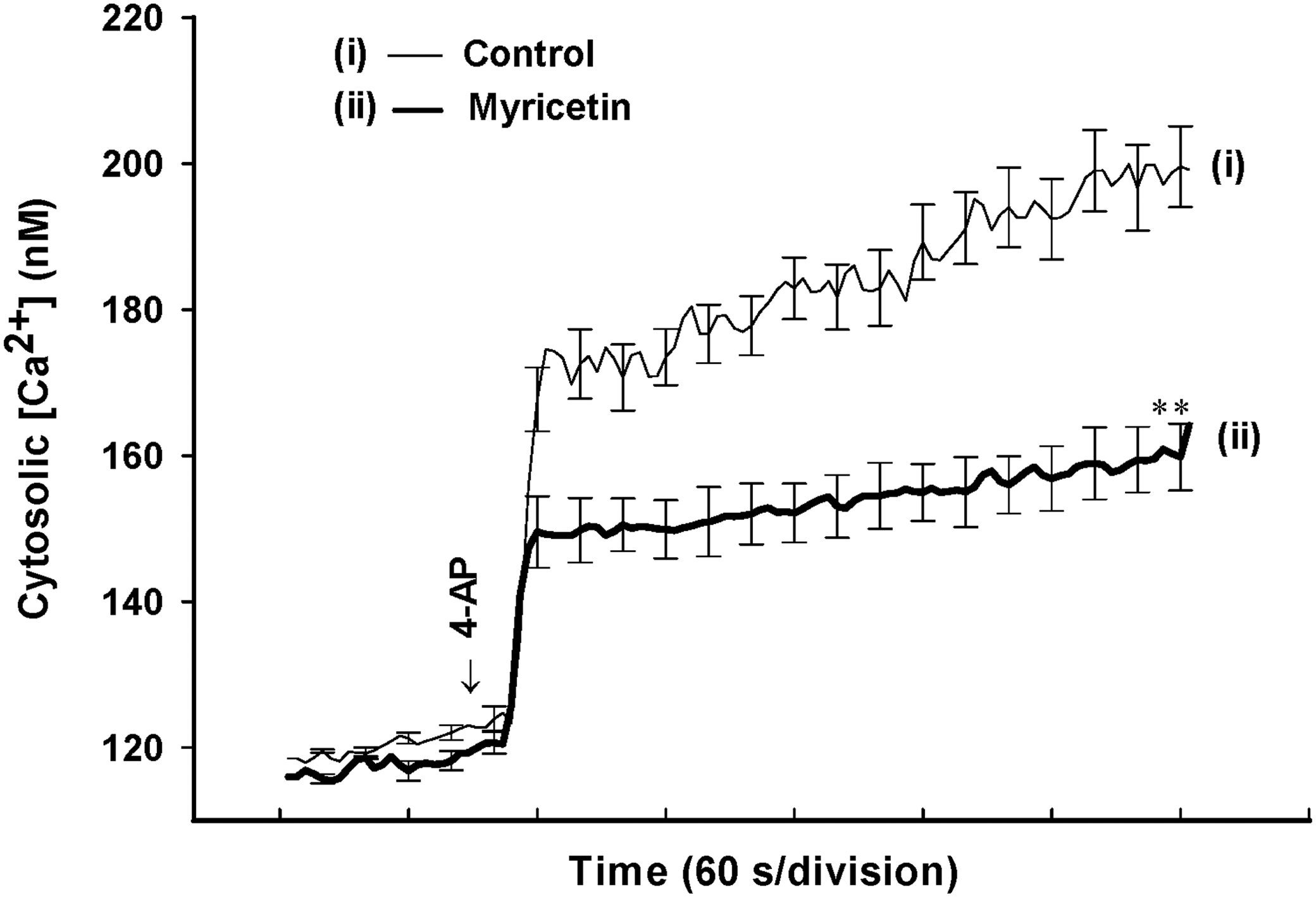
4-AP-evoked increase in cytosolic Ca2+ concentrations ([Ca2+]C) is reduced by myricetin. Cytosolic free Ca2+ concentration (nM) was monitored using Fura-2 in the absence (control) or presence of 30 μM myricetin added 10 min before depolarization with 1 mM 4-AP. Results are mean±SEM of five independent experiments. **P<.01 versus control.
In the adult rat cerebrocortical nerve terminal preparation, the release of glutamate evoked by depolarization is reported to be caused by Ca2+ influx through Cav2.2 (N-type) and Cav2.1 (P/Q-type) channels and Ca2+ release from internal stores such as the endoplasmic reticulum (ER) and mitochondria. 33 –35 Therefore, we examined which part of the Ca2+ source was involved in the myricetin-mediated inhibition of glutamate release. In Figure 5A, ω-conotoxin MVIIC (2 μM), a wide-spectrum blocker of Cav2.2 and Cav2.1 channels, reduced 4-AP (1 mM)-evoked glutamate release from 7.3±0.3 nmol/mg/5 min to 2.4±0.3 nmol/mg/5 min (32.1±4.2% control; P<.01). In the presence of ω-CgTX MVIIC (2 μM), the inhibitory effect of myricetin on 4-AP-evoked glutamate release was significantly prevented (Fig. 5A, D). In addition, dantrolene (50 μM), an inhibitor of intracellular Ca2+ release from ER, reduced the 4-AP (1 mM)-evoked glutamate release (n=7; P<.05; Fig. 5B), indicating that Ca2+ release from ER ryanodine receptors contributes significantly to the 4-AP-evoked glutamate release. In the presence of dantrolene, myricetin (30 μM) was able to produce a 61.5±4.2% inhibition of 4-AP-evoked glutamate release, which was similar to that observed for the 56.7±3.7% inhibition produced by myricetin (30 μM) alone (n=7; Fig. 5B, D). Similar results were observed with 100 μM CGP37157, a membrane-permeable blocker of mitochondrial Na+/Ca2+ exchange (n=5; Fig. 5C, D). These results suggest that decreases in the Ca2+ release from intracellular stores do not mediate the inhibitory effect of myricetin on glutamate release.

Myricetin-mediated inhibition of 4-AP-evoked glutamate release is prevented by blocking the Cav2.2 and Cav2.1 channels. Glutamate release was evoked by 1 mM 4-AP in the absence (control) or presence of 2 μM ω-conotoxin MVIIC (ω-CgTX MVIIC)
Discussion
By preparing nerve terminals (synaptosomes) from rat cerebral cortex, this study is the first to report that myricetin preferentially inhibits depolarization-evoked glutamate release. Several possible mechanisms for this effect are discussed as follows.
The process through which neurotransmitter release occurs is complex, involving Na+, K+, and Ca2+ channels.
36
The inhibition of Na+ channels and the activation of K+ channels stabilizes membrane excitability and, consequently, causes a decrease in the levels of Ca2+ entry and neurotransmitter release.
37
–39
Therefore, addressing the mechanism responsible for the myricetin-mediated inhibition of glutamate release, this study considers two scenarios that might be involved: (1) alteration of the synaptosomal plasma membrane potential and (2) direct regulation of VDCCs affecting Ca2+ entry. The first possibility is unlikely because of the following observations. First, 4-AP- and KCl-evoked glutamate release was significantly inhibited by myricetin. Because 4-AP-evoked glutamate release involves the activation of Na+ and Ca2+ channels, 15 mM external KCl-evoked glutamate release involves only Ca2+ channels
30,32
; thus, Na+ channels are not involved in the effect of myricetin on glutamate release. Second, myricetin was not observed to substantially affect the synaptosomal plasma membrane potential, indicating that myricetin does not affect K+ conductance. Third, myricetin did not affect the 4-AP-evoked Ca2+-independent nonvesicular glutamate release, a component of glutamate release that is exclusively dependent on membrane potential.
31
This indicates that myricetin does not affect glutamate release by reversing the direction of the plasma membrane glutamate transporter. In addition, the inhibitory effect of myricetin on 4-AP-evoked glutamate release was prevented by the vesicular transporter inhibitor bafilomycin A1, but not by the glutamate transporter inhibitor
In synaptic terminals, extracellular Ca2+ influx through VDCCs and intracellular store Ca2+ release mediate a depolarization-induced increase in [Ca2+]C coupled with glutamate release. 33 –35 Using the Ca2+ indicator Fura-2, this study demonstrates that myricetin reduces the 4-AP-evoked increase in [Ca2+]C. Furthermore, the inhibition of glutamate release produced by myricetin was prevented when Cav2.2 and Cav2.1 channels were blocked. Neither dantrolene, an inhibitor of intracellular Ca2+ release from the ER, nor CGP37157, a mitochondrial Na+/Ca2+ exchange blocker, affected the inhibitory effect of myricetin on 4-AP-evoked glutamate release. Thus, the reduced release of stored Ca2+ from the ER ryanodine receptors and mitochondria during the myricetin-mediated inhibition of glutamate release can be excluded. Although no direct evidence has indicated that myricetin acts on presynaptic Ca2+ channels, these data implied that the suppression of Ca2+ influx through Cav2.2 and Cav2.1 channels is involved in the myricetin-mediated inhibition of glutamate release. Our finding, although contrasting with some previous studies, 43 is consistent with previous studies demonstrating that myricetin inhibits Ca2+ channels in several experimental models. 42,44 Besides a participation of presynaptic Ca2+ channel in the inhibitory effect of myricetin on glutamate release, the possible involvement of other presynaptic pathways should be considered. For example, myricetin has been reported to enhance the GABAergic activity in rat brain slices and neuronal cultures. 43 Future studies are needed to determine whether a relationship exists between the inhibitory effect of myricetin on glutamate release and the GABA-enhancing effect of myricetin.
Recently, numerous studies have focused on several flavonoids that have been suggested to exhibit potential therapeutic applications for treating neurological disorders. For example, quercetin has been confirmed to penetrate the blood–brain barrier
45
and protect against ischemia-induced neuronal damage and cognitive dysfunction.
46
The structure of myricetin is similar to that of quercetin. Although no study has established the brain levels of myricetin after systemic injection in rats, administering myricetin to animals at a dose of 100 mg/kg (∼ 500 μM) ameliorates
In conclusion, the results of the current study indicate that myricetin inhibits glutamate release from rat cortical nerve terminals by suppressing presynaptic Ca2+ influx. As such, this represents an important mechanism of action of myricetin, which might contribute to its prevention of calcium overload associated with neuronal injury.
Footnotes
Acknowledgment
This work was supported by a grant from the Shin Kong Wu Ho-Su Memorial Hospital (101-SKH-FJH-04), Taiwan.
Author Disclosure Statement
The authors affirm that no competing financial interests exist.