
Abstract
In the current study, we evaluated the anti-inflammatory effects of Lonicera japonica THUNB. (LJ) and its underlying molecular mechanism in lipopolysaccharide (LPS)-stimulated BV-2 microglial cells. Our results indicated that LJ significantly inhibits LPS-stimulated production of nitric oxide (NO) and prostaglandin E2 (PGE2). In addition, LJ inhibited inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) at both the protein and mRNA levels. In LPS-stimulated BV-2 microglial cells, LJ inhibited proinflammatory cytokines and chemokines, tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), monocyte chemoattractant protein-1 (MCP-1), matrix metalloproteinase-9 (MMP-9) enzymatic activities, and/or mRNA expression, as well as reactive oxygen species (ROS) production. LJ significantly suppressed activation of nuclear factor-κB (NF-κB) and its translocation from the cytosol to the nucleus and suppressed the DNA-binding activity of NF-κB. Furthermore, LJ significantly inhibited phosphorylation of c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase 1/2 (ERK 1/2), p38 mitogen-activated protein kinases (MAPKs), phosphatidylinositol 3-kinases (PI3K)/Akt, and Janus kinase 1 (JAK1)/signal transducer and activator of transcription (STAT)1/3. Collectively, our findings indicated that the antineuroinflammatory properties of LJ in LPS-induced BV-2 microglial cells is due to downregulation of proinflammatory cytokines and chemokines downstream of inhibition of NF-κB activation.
Introduction
M
Lipopolysaccharide (LPS) is a bacterial endotoxin used to study experimentally induced infection, inflammation, and tissue damage, as well as the biochemistry of inflammatory responses. LPS activates ROS, nuclear factor-κB (NF-κB), phosphatidylinositol 3-kinases (PI3K)/Akt, and members of the mitogen-activated protein kinase (MAPK) family, which are classified into at least three components: c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase 1/2 (ERK 1/2), and p38 MAPK. 8 –10 Moreover, to activate neuroinflammatory signaling in LPS-induced microglial cells, LPS also activates the Janus kinase1 (Jak1)/signal transducer and activator of transcription (STAT)1/3 signaling pathway and is important for cytokine and chemokine production. 11 All of these factors have been implicated in the release of immune-related cytotoxic factors, such as iNOS, cyclooxygenase-2 (COX-2), NO, and proinflammatory cytokines, either separate from or in addition to chemokines, including TNF-α, IL-1β, MCP-1, and MMP-9. 11 –13
Lonicera japonica THUNB. (LJ, honeysuckle) is a widely used traditional Korean natural herb with a variety of biological functions and therapeutic properties, including antiswelling, antiviral relief, antitumor, antipyretic, antiapoptotic, anti-inflammatory, and antibacterial effects. 14 –17 Traditional claims and pharmacological studies have indicated that LJ may be an attractive candidate for the treatment of various CNS disorders and neurodegenerative diseases. Recently, ethanolic extracts of LJ were shown to inhibit aggregation and neurotoxicity of amyloid beta1–42 (Aβ 1–42) in human neuroblastoma cells. 18 In addition, it was previously reported that methanolic extracts of LJ have neuroprotective activity against glutamate-induced neurotoxicity in primary cultured rat cortical cells. 19 In our previous studies, we also demonstrated the effect of LJ in promoting oxidative stress-induced neuronal cell death through activation of MAPKs, PI3K/Akt, and NF-κB in SH-SY5Y cellular models. 20,21 Recently, it was reported that LJ has anti-inflammatory properties in animal models of osteoarthritis and excision wounding. 22,23 For example, in these animal models, LJ was found to suppress a number of inflammation-related phenomena and the amount of cytokines produced, including TNF-α and IL-6. Moreover, LJ was shown to have anti-inflammatory effects in a carrageenan-induced paw edema inflammation model. Although the anti-inflammatory effects of LJ in these animal models have been well documented, its effects on cells in the CNS, specifically the microglia, remain elusive. Indeed, the detailed molecular mechanisms underlying the effects of LJ on neuroinflammation have not been investigated. Therefore, in this study, we investigated the pharmacological effects of LJ on microglia activated by LPS. In addition, we examined whether LJ has antineuroinflammatory activity through upregulation of iNOS, COX-2, MMP-9, proinflammatory cytokines, and chemokines, as well as ROS accumulation in LPS-stimulated BV-2 microglial cells. We also assessed LJ's anti-inflammatory properties and determined if LJ reduces inflammation by inhibiting phosphorylation of MAPKs, PI3K/Akt, and JAK1/STAT1/3, as well as activation of NF-κB.
Materials and Methods
Materials
2,7′-Dichlorofluorescin diacetate (DCFH-DA), dimethyl sulfoxide (DMSO), Hoechst 33258, 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), LPS (Escherichia coli, 026:B6), poly-D-lysine, and anti-β-actin antibody were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM) was obtained from Hyclone (Logan, UT, USA). Fetal bovine serum (FBS), 0.25% trypsin-EDTA, and penicillin/streptomycin were obtained from GIBCO–BRL (Grand Island, NY, USA). Rabbit anti-phospho-Akt (Ser473), rabbit anti-rabbit anti-phospho-ERK1/2 (Thr202/Tyr204), rabbit anti-ERK 1/2 (Thr202/Tyr204), rabbit anti-phospho-JAK1 (Tyr1022/1023), rabbit anti-JAK1, rabbit anti-phospho-JNK (Thr183/Tyr185), rabbit anti-JNK (Thr183/Tyr185), rabbit anti-lamin B1, rabbit anti-NF-κB p65, rabbit anti-phospho-STAT1 (Tyr701), rabbit anti-STAT1, rabbit anti-phospho-STAT3 (Tyr705), rabbit anti-STAT3, and anti-rabbit horseradish peroxidase-linked IgG antibodies were purchased from Cell Signaling (Boston, MA, USA). Rabbit anti-COX-2, rabbit anti-phospho-IκB-α, rabbit anti-IκB-α, rabbit anti-phospho-p38 MAPK (Thr180/Tyr182), and rabbit anti-p38 MAPK (Thr180/Tyr182) antibodies were purchased from Epitomics (Burlingame, CA, USA). Rabbit anti-iNOS and rabbit anti-Akt (Ser473) antibodies were obtained from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA). Texas red®-conjugated goat anti-rabbit IgG antibody and lipofectamine® 2000 transfection reagent were purchased from Invitrogen (Carlsbad, CA, USA). Polymerase chain reaction (PCR) primers were synthesized from Cosmogenetech Co., Ltd., (Seoul, Korea). All other chemicals were of analytical grade from Sigma Chemical Co.
Preparation of LJ extract
Dried LJ flower buds were purchased from the Jung-Do Herbal Drug Company (Seoul, Korea). The dried LJ flower buds were collected in Goseong, Gangwon-do, South Korea, in early June 2008, and were identified by Professor Sun Yeou Kim (College of Pharmacy, Gachon University, Incheon, Korea). The dried LJ flower buds were cut into small pieces and extracted thrice with 1 kg/L of distilled water at 70°C with a cooling system (40°C) for 3 h. The water extract was filtered through Whatman No. 2 filter paper, 10 and the supernatant was concentrated under reduced pressure in a vacuum rotary evaporator (N-1000; EYELA, Tokyo, Japan). Finally, the supernatant (400 g) was extracted with ethyl acetate thrice for 1 h in an ultrasonic apparatus. The supernatant was evaporated and spray-dried to yield 12 g of LJ extract.
Cell culture and treatment
BV-2 microglial cells were grown in DMEM supplemented with 10% heat-inactivated FBS (v/v) and 0.1% penicillin/streptomycin (v/v) in a humidified atmosphere of 5% CO2 and 95% air at 37°C. LPS was prepared immediately before use as 10 μg/mL stock and diluted in phosphate-buffered saline (PBS) to the indicated final concentration. LJ was dissolved in DMSO and the stock solutions were added directly to the culture media. Control cells were treated with DMSO only. The final concentration of solvent was always <0.1% (v/v). No significant cytotoxicity was observed in any of the experiments (data not shown). In all experiments, cells were treated with the indicated concentrations of LJ in the presence or absence of LPS (100 ng/mL) in serum-free DMEM.
Determination of NO production
NO release in the culture supernatants was measured by the Griess reaction. In brief, BV-2 microglial cells (2.5×105 cells/well in 24-well plates) were incubated at 37°C with LPS for 24 h with or without LJ pretreatment, and the supernatant was assayed. Then, 100 μL of culture supernatant from each sample was mixed with the same volume of Griess reagent [0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride and 1% sulfanilamide in 5% phosphoric acid] in 96-well plates for 10 min at room temperature in the dark. Nitrite concentrations were determined using standard solutions of sodium nitrite prepared in cell culture medium. The absorbance at 540 nm was determined using a microplate reader (SpectraMax 250; Molecular Device, Sunnyvale, CA, USA).
Enzyme-linked immunosorbent assay
Following the manufacturer's instructions, levels of PGE2 (Cayman Chemical, Ann Arbor, MI, USA), TNF-α, IL-1β, MCP-1 from KOMA Biotech, Inc. (Seoul, Korea), and MMP-9 (R&D Systems, Inc., Minneapolis, MN, USA) were determined using enzyme-linked immunosorbent assay (ELISA) kits.
Measurement of intracellular ROS accumulation
BV-2 microglial cells (1×106 cells/well) were seeded on six-well plates or poly-D-lysine-coated slides overnight. Intracellular ROS levels were examined using DCFH-DA as previously described. 24,25 After pretreatment with or without LJ for 30 min, the cells were incubated with LPS for 24 h. The cells were then rinsed with PBS and 10 μM DCFH-DA was added for 30 min at 37°C, washed twice with PBS, and examined at 530 nm with a fluorescence microplate reader (SpectraMax M2; Molecular Device) with excitation at 488 nm. DCFH-DA fluorescence images were collected using a fluorescence microscope (20×).
RNA isolation and reverse transcription–polymerase chain reactions
BV-2 microglial cells (1×106 cells/well in six-well plates) were incubated at 37°C with LPS for 6 h with or without LJ pretreatment. Total RNA was isolated using Trizol® reagent (Invitrogen). Reverse transcription reactions were carried out with the Superscript®-III kit (Invitrogen) using 5 μg of total RNA and oligo dT according to the manufacturer's instructions. PCR products were analyzed by staining with ethidium bromide on 1.5% agarose gels in Tris borate/EDTA buffer (890 mM Tris-Base, 890 mM boric acid, 20 mM EDTA, pH 8.3) after electrophoresis for 30 min at 100 V. Band intensities of the amplified DNAs were compared after visualization on a UV transilluminator. mRNA bands were quantified by densitometric analysis using ImageJ software (NIH Image in the public domain, USA). Specific primer sequences are described in Supplementary Table S1 (Supplementary Data are available online at
Nuclear and cytosolic lysate preparation
BV-2 microglial cells were seeded at a density of 5×106 cells/well in 100-mm2 cell culture dishes. After pretreatment with LJ for 30 min, the cells were incubated with LPS for 1 h. To measure activation of NF-κB p65 in the nucleus, nuclear and cytosolic fractions were prepared using NE-PER nuclear and cytoplasmic extraction reagents for cultured cells (Pierce, Rockford, IL, USA) according to the manufacturer's instructions. NF-κB p65 levels were determined by western blot analysis and electrophoretic mobility shift assay (EMSA) as described below.
Western blot analysis
BV-2 microglial cells were seeded at a density of 1×106 cells/well in six-well plates. After pretreatment with LJ for 30 min, the cells were incubated with LPS for 1, 2, or 24 h. Next, cells were washed with ice-cold PBS and harvested by scraping with 100 μL of ice-cold lysis T-per tissue protein extraction buffer (Thermo Scientific, Rockford, IL, USA) containing protease and phosphatase inhibitor cocktails (Roche Diagnostics GmbH, Mannheim, Germany). The lysates were then incubated on ice for 30 min. After centrifugation at 10,000 g for 15 min, supernatants were separated and stored at −70°C. Protein concentrations were determined using a protein assay kit (Thermo Scientific). Cell lysates were separated on 8–12% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride transfer membranes (Pall Corporation, Pensacola, FL, USA), Next, the membranes were blocked with 5% skim milk containing TBST buffer (0.5 mM Tris–HCl [pH 7.5], 150 mM NaCl, and 0.1% Tween-20) for 1 h at room temperature. The membranes were subsequently incubated with primary antibody overnight at 4°C [each antibody at a dilution of 1:1000; iNOS, phospho-JNK (Thr183/Tyr185), JNK, phospho-ERK1/2 (Thr202/Tyr204), ERK 1/2 (Thr202/Tyr204), phospho-JAK1 (Tyr1022/1023), JAK1, phospho-Akt (Ser473), Akt (Ser473), NF-κB p65, phospho-STAT1 (Tyr701), STAT1, phospho-STAT3 (Tyr705), STAT3, and lamin B1, except β-actin (1:20,000), COX-2 (1:500), phospho-IκB-α (1:10,000), and IκB-α (1:10,000)]. After three washes with TBST, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies in TBST with 5% nonfat milk at a 1:5000 dilution for 1 h at room temperature. The blots were then washed thrice in TBST buffer. Blots were developed using the enhanced chemiluminescence detection method by immersing them for 5 min in a mixture of ECL reagents (Animal Genetics, Inc., Suwon, Korea) A and B at a 1:1 ratio and exposing to photographic film for a few minutes. Protein bands were quantified by densitometric analysis using ImageJ software.
Transient transfection and dual-luciferase assay
The NF-κB reporter construct used in this study was purchased from SABiosiences, Inc., (QIAGEN, Inc., Valencia, CA, USA). Briefly, BV-2 microglial cells were plated onto 24-well plates at a density of 2.5×105 cells/well and grown overnight. Cells were cotransfected with 5 μg/mL of the NF-κB plasmid construct or negative plasmid constructs using lipofectamine. An internal control was also transfected to measure transfection efficiency after 6 h. After transfection, cells were cultured in 10% FBS medium for 24 h. Twenty-four hours after transfection, cells were incubated with LJ for 1 h, followed by LPS for 6 h. Luciferase activity was assayed using a dual-luciferase assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. Luminescence was measured using a single-tube luminometer (FB12; Berthold Detection Systems GmbH, Pforzheim, Germany).
Electrophoretic mobility shift assay
Nuclear extracts were prepared using NE-PER nuclear and cytoplasmic extraction reagents as described above. Synthetic complementary NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′)-binding oligonucleotides (Panomics, Inc., Santa Clara, CA, USA) were 5′-biotinylated using a biotin 5′-end DNA-labeling EMSA kit according to the manufacturer's protocol (Affymetrix, Inc., Santa Clara, CA, USA). Binding reactions contained 10 μg of nuclear extract protein, binding buffer, 1 μg of poly d(I-C), and 10 ng of biotin-labeled DNA. The reactions were incubated for 5 min at room temperature in a final volume of 10 μL. The protein-DNA complex was separated from the DNA probe by electrophoresis on native 6% polyacrylamide gels pre-electrophoresed for 1 h in 0.5× Tris borate/EDTA buffer (50 mM Tris-Base, 18 mM boric acid, 500 mM EDTA, pH 8.3) before being transferred onto a positively charged nylon membrane (Pall Corporation) in 0.5× Tris borate/EDTA buffer at 300 mA for 30 min. Next, the transferred DNAs were cross-linked to the nylon membrane in a dry oven at 80°C for 1 h. Horseradish peroxidase-conjugated streptavidin was used according to the manufacturer's instructions to detect the transferred DNAs.
Immunocytochemistry
BV-2 microglial cells (2.5×105 cells/well) were seeded on culture slides for 24 h. After pretreatment with LJ for 30 min, the cells were incubated with LPS for 1 h. Then, cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min. After washing, cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min. Cells were then blocked in a 5% bovine serum albumin solution in PBS for 1 h, followed by incubation with anti-NF-κB p65 (1:250) overnight. Next, cells were washed with PBS and incubated for 1 h with Texas red-conjugated goat anti-rabbit IgG antibody (1:500) and Hoechst 33258 (5 μg/mL) for 5 min. Cells were washed in PBS and mounted on glass slides in Permafluor aqueous mounting fluid. All procedures were performed at room temperature. Cells were observed under a fluorescence microscope (100×). Results are representative of three independent experiments.
Statistics
Data were analyzed with Prism 5.0 software (Graphpad Software, Inc., San Diego, CA, USA) and expressed as the mean±SEM. Statistical analyses were performed using one-way analysis of variance followed by the Newman–Keuls test. Statistical significance was set at P<.05.
Results
Effects of LJ on cell viability in BV-2 microglial cells
To exclude the possibility that the decrease in NO and cytokine levels was simply due to cell death, the cytotoxic effects of the LJ extract in BV-2 microglial cells were evaluated in the absence or presence of LPS using MTT assays. The MTT assays showed that LJ was not cytotoxic at the concentrations (0.5, 5, 2.5, 5, and 10 μg/mL) used in this study (Supplementary Fig. S1A, B). Thus, these concentrations of LJ were deemed appropriate and used in subsequent experiments.
LJ inhibits the production of NO and PGE2 in LPS-stimulated BV-2 microglial cells
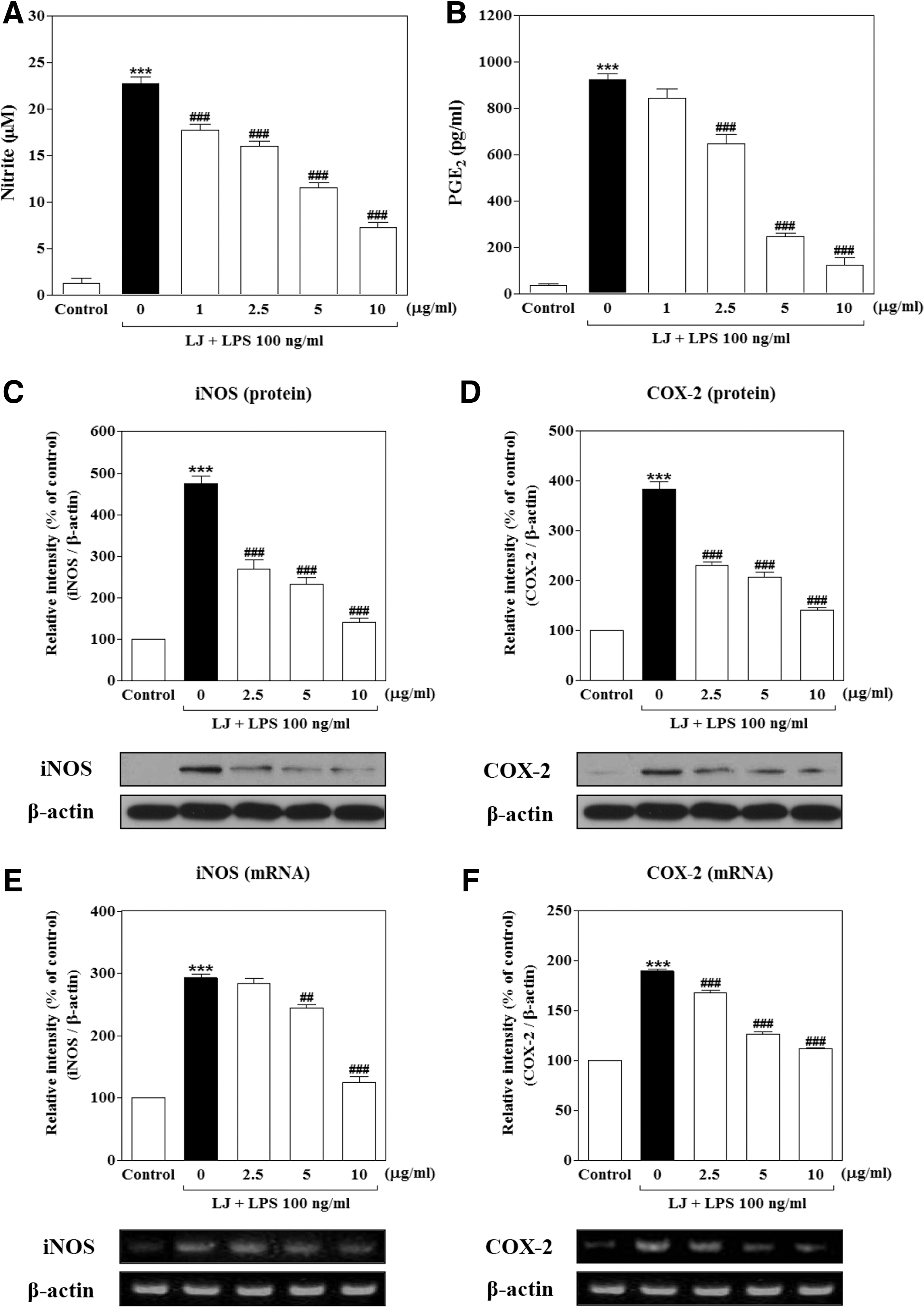
Initially, we determined the effects of LJ on NO and PGE2 production in LPS-stimulated BV-2 microglial cells. To determine NO production, we measured the level of nitrite released into the culture medium using Griess reagent. Treatment with LPS significantly increased NO and PGE2 production to 22.72±0.74 μM and 922.60±25.72 pg/mL of the control values, respectively (Fig. 1A, B, P<.001). However, this increased production of NO was significantly inhibited by 1 μg/mL of LJ to 17.72±0.66 μM of the control value (P<.001), and 2.5 μg/mL of LJ significantly reduced production of NO and PGE2 to 16.00±0.54 μM and 648.50±38.26 pg/mL, respectively (P<.001). Pretreatment with 5 μg/mL of LJ also decreased NO and PGE2 production to 11.50±0.56 μM and 248.00±14.41 pg/mL, respectively (P<.001 compared with controls). In addition, pretreatment with 10 μg/mL of LJ dramatically inhibited NO and PGE2 production to 7.22±0.54 μM and 124.00±32.13 pg/mL, respectively (P<.001 compared with control values).
LJ inhibits the production of NO
LJ attenuates LPS-induced expression of iNOS and COX-2 protein and mRNA levels in BV-2 microglial cells
Because LJ was found to inhibit NO and PGE2 production, we next examined the relationship between LJ levels and the expression of iNOS and COX-2. Western blot analysis showed that treatment with LPS significantly increased expression of iNOS and COX-2 protein levels to 475.00%±17.19% and 384.00%±14.59% of the control values, respectively (Fig. 1C, D, P<.001). However, upregulation of iNOS and COX-2 protein levels was significantly attenuated by 2.5 μg/mL of LJ to 268.20%±22.23% and 230.00%±6.87% of the control values (P<.001), whereas upregulation of iNOS and COX-2 protein levels was decreased to 233.00%±14.39% and 207.00%±9.78% of the control values at 5 μg/mL, respectively (P<.001). In addition, pretreatment with 10 μg/mL of LJ markedly inhibited upregulation of iNOS and COX-2 protein levels to 141.20%±8.43% and 140.50%±5.49% of the control values, respectively (P<.001).
Treatment with LPS also significantly increased the expression of iNOS and COX-2 mRNA levels to 292.50%±6.23% and 189.60%±2.21% of the control values, respectively (Fig. 1E, F, P<.001). However, upregulation of the COX-2 mRNA level was significantly inhibited by 2.5 μg/mL of LJ to 168.20%±2.63% of the control value (P<.001), whereas upregulation of iNOS and COX-2 levels was decreased to 243.90%±6.05% and 126.70%±1.97% of the control values with 5 μg/mL of LJ, respectively (P<.01 and P<.001). In addition, pretreatment with 10 μg/mL of LJ significantly inhibited upregulation of iNOS and COX-2 mRNA levels to 125.10%±9.03% and 112.40%±0.63% of the control values, respectively (P<.001).
LJ inhibits LPS-induced TNF-α, IL-1β, MCP-1, and MMP-9 production in BV-2 microglial cells
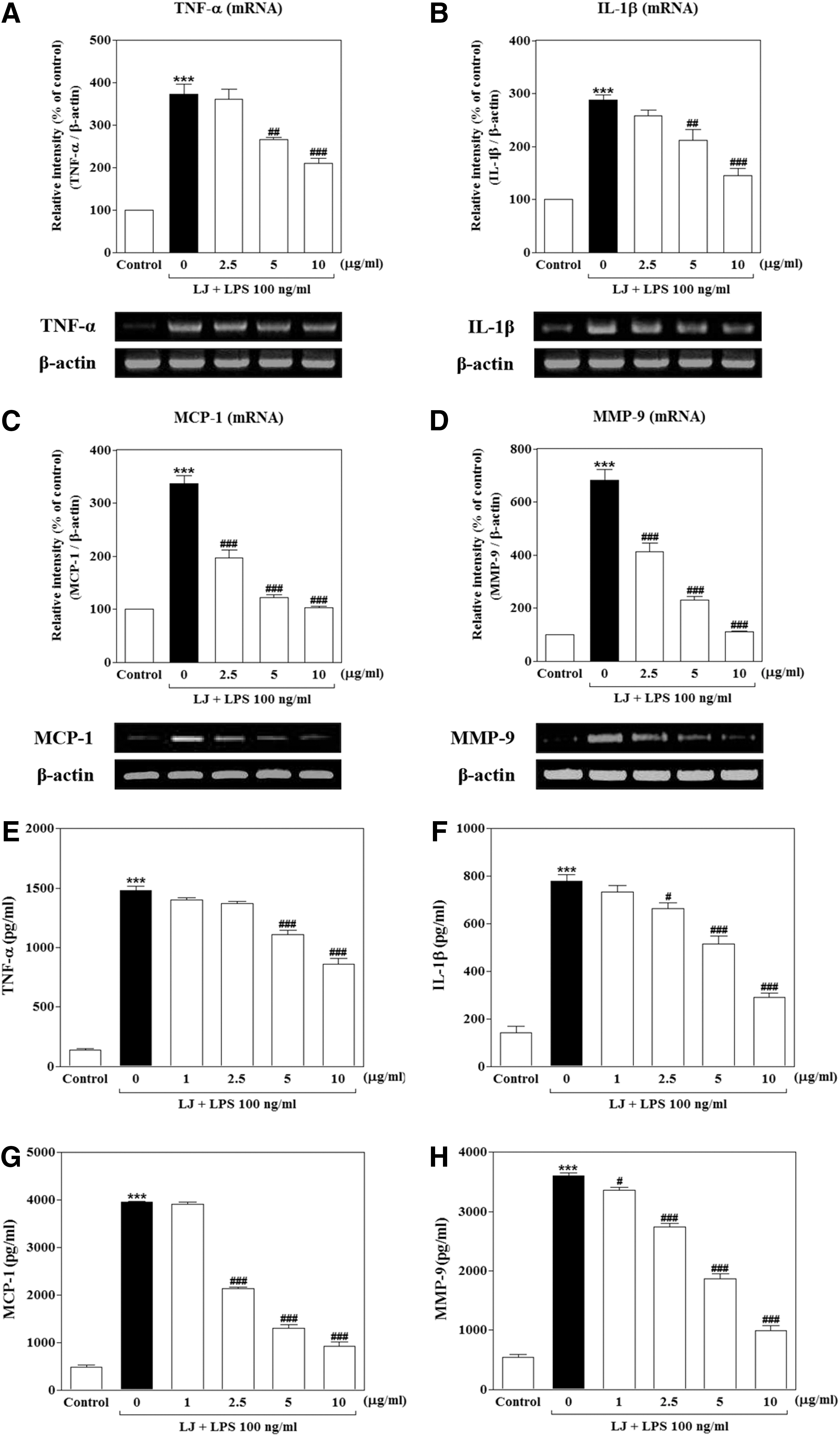
We investigated whether LJ inhibits LPS-induced production of TNF-α, IL-1β, MCP-1, and MMP-9 production using reverse transcription–polymerase chain reactions (RT-PCR) and ELISA. Treatment with LPS significantly increased the mRNA levels of TNF-α, IL-1β, and MCP-1 proinflammatory cytokines or chemokine to 372.90%±23.88%, 287.90%±9.39%, and 336.80%±15.41% of the control values, respectively (Fig. 2A–C, P<.001), and the mRNA levels of MMP-9 were increased to 682.40%±39.63% of the control value (Fig. 2D, P<.001). On the other hand, upregulation of MCP-1 and MMP-9 mRNA levels was significantly suppressed by 2.5 μg/mL of LJ to 196.50%±14.59% and 413.30%±32.45% of the control values, respectively (P<.001). Pretreatment with 5 μg/mL of LJ also significantly attenuated upregulation of TNF-α, IL-1β, MCP-1, and MMP-9 mRNA levels to 265.90%±6.31%, 211.20%±21.58%, 122.10%±5.99%, and 229.90%±14.09% of the control values, respectively (P<.01 and P<.001). In addition, pretreatment with 10 μg/mL of LJ markedly inhibited upregulation of TNF-α, IL-1β, MCP-1, and MMP-9 mRNA levels to 210.80%±11.42%, 144.70%±13.42%, 102.80%±3.50%, and 110.30%±2.53% of the control values, respectively (P<.001). In a parallel approach, we performed ELISA to determine whether LJ inhibited release of these factors at detectable levels under culture medium conditions. Treatment with LPS significantly increased TNF-α, IL-1β, MCP-1, and MMP-9 secretion to 1478.00±36.86, 778.20±27.01, 3958.00±15.99, and 3604.00±45.25 pg/mL, respectively (Fig. 2E–H, P<.001). However, secretion of MMP-9 was significantly decreased following treatment with 1 μg/mL of LJ to 3353.00±55.25 pg/mL (P<.05), and pretreatment with 2.5 μg/mL of LJ also decreased IL-1β, MCP-1, and MMP-9 secretion to 662.50±26.46, 2142.00±27.04, and 2739.00±55.95 pg/mL, respectively (P<.05 and P<.001). Pretreatment with 5 μg/mL of LJ also significantly decreased TNF-α, IL-1β, MCP-1, and MMP-9 secretion to 1109.00±37.15, 515.00±33.77, 1307.00±69.99, and 1870.00±74.56 pg/mL, respectively (P<.001). Last, pretreatment with 10 μg/mL of LJ markedly inhibited TNF-α, IL-1β, MCP-1, and MMP-9 secretion to 860.30±47.22, 291.80±18.17, 920.60±96.29, and 986.10±95.32 pg/mL, respectively (P<.001).
LJ inhibits LPS-induced TNF-α
LJ inhibits LPS-induced ROS accumulation in BV-2 microglial cells
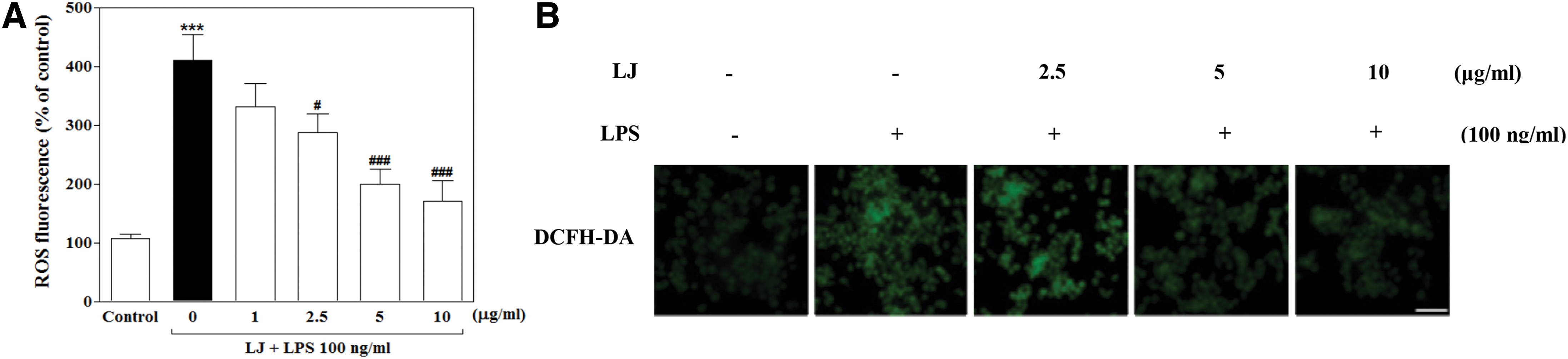
We next examined the effect of LJ on ROS, which are known to be early inducers of inflammation. ROS are produced by microglia and contribute to neuronal cell death and neurodegeneration. In this study, we investigated intracellular ROS formation using DCFH-DA, a fluorescent ROS-sensitive probe. Treatment with LPS significantly increased intracellular ROS production to 410.00%±44.07% of the control value (Fig. 3A, P<.001), whereas pretreatment with 2.5, 5, and 10 μg/mL of LJ significantly inhibited this increase in intracellular ROS accumulation to 287.80%±31.47%, 199.80%±25.12%, and 171.40%±34.95% of the control values, respectively (P<.05 and P<.001). To further investigate the effect of LJ on LPS-induced intracellular ROS accumulation, we performed immunostaining using the DCFH-DA probe. Microphotographs of DCFH-DA staining revealed excessive intracellular ROS accumulation after LPS stimulation (Fig. 3B). Interestingly, pretreatment with LJ clearly inhibited signaling events leading to intracellular ROS accumulation.
LJ inhibits LPS-induced ROS accumulation in BV-2 microglial cells
LJ suppresses LPS-induced phosphorylation of JNK, ERK 1/2, p38 MAPKs, and PI3K/Akt in BV-2 microglial cells
To evaluate the effects of LJ on the upstream signaling pathways associated with activation of NF-κB translocation, we examined changes in the activation of intracellular signaling proteins, such as JNK, ERK 1/2, p38 MAPKs, and PI3K/Akt in BV-2 microglial cells. As shown in Figure 5, treatment with LPS dramatically and rapidly increased the phosphorylation of JNK, ERK 1/2, p38 MAPKs, and PI3K/Akt to 466.60%±17.04%, 277.80%±21.95%, 179.00%±13.54%, and 185.50%±8.12% of the control values, respectively (Fig. 4A–D, P<.001). However, the increase of phosphorylation of p38 MAPK and Akt was significantly inhibited by 5 μg/mL of LJ to 133.50%±3.38% and 144.30%±9.01% of the control values, respectively (P<.05 and P<.01). Moreover, pretreatment with 10 μg/mL of LJ significantly suppressed phosphorylation of JNK, ERK 1/2, p38 MAPKs, and PI3K/Akt to 265.60%±9.84%, 120.80%±7.80%, 115.10%±6.14%, and 113.40%±2.40% of the control values, respectively (P<.05 and P<.001).

LJ suppresses LPS-induced phosphorylation of JNK

LJ suppresses LPS-induced phosphorylation of JAK1
LJ suppresses LPS-induced phosphorylation of JAK1 and STAT1/3 in BV-2 microglial cells
Because LJ reduced the activation of neuroinflammatory molecules, it likely blocks signaling and transcriptional events downstream of neuroinflammatory target genes. In addition to activation of NF-κB translocation in LPS-induced BV-2 microglial cells, LPS also activates the JAK1 and STAT1/3 signaling pathways, which are important for cytokine and/or chemokine production. Thus, we investigated the effects of LJ on LPS-induced phosphorylation of JAK1 and STAT1/3 activation. Evaluation of the phosphorylation status of JAK1 and STAT1/3 revealed that maximum phosphorylation levels occurred 2 h after LPS stimulation (data not shown). Treatment with LPS significantly increased the phosphorylation of JAK1, STAT1, and STAT3 to 323.70%±5.43%, 433.60%±24.01%, and 487.20%±17.50% of the control values, respectively (Fig. 5A–C, P<.001). The increase in STAT1 phosphorylation was significantly inhibited by 2.5 μg/mL of LJ to 351.80%±39.26% of the control value. Pretreatment with 5 μg/mL of LJ also significantly suppressed the phosphorylation of JAK1, STAT1, and STAT3 to 284.20%±7.07%, 266.40%±32.03%, and 349.20%±15.27% of the control values, respectively (P<.05, P<.01, and P<.001). Last, pretreatment with 10 μg/mL LJ significantly inhibited the phosphorylation of JAK1, STAT1, and STAT3 to 160.60%±10.51%, 171.90%±8.60%, and 299.90%±10.58% of the control values, respectively (P<.001).
LJ inhibits both LPS-induced phosphorylation and degradation of IκB-α as well as activation of NF-κB in BV-2 microglial cells
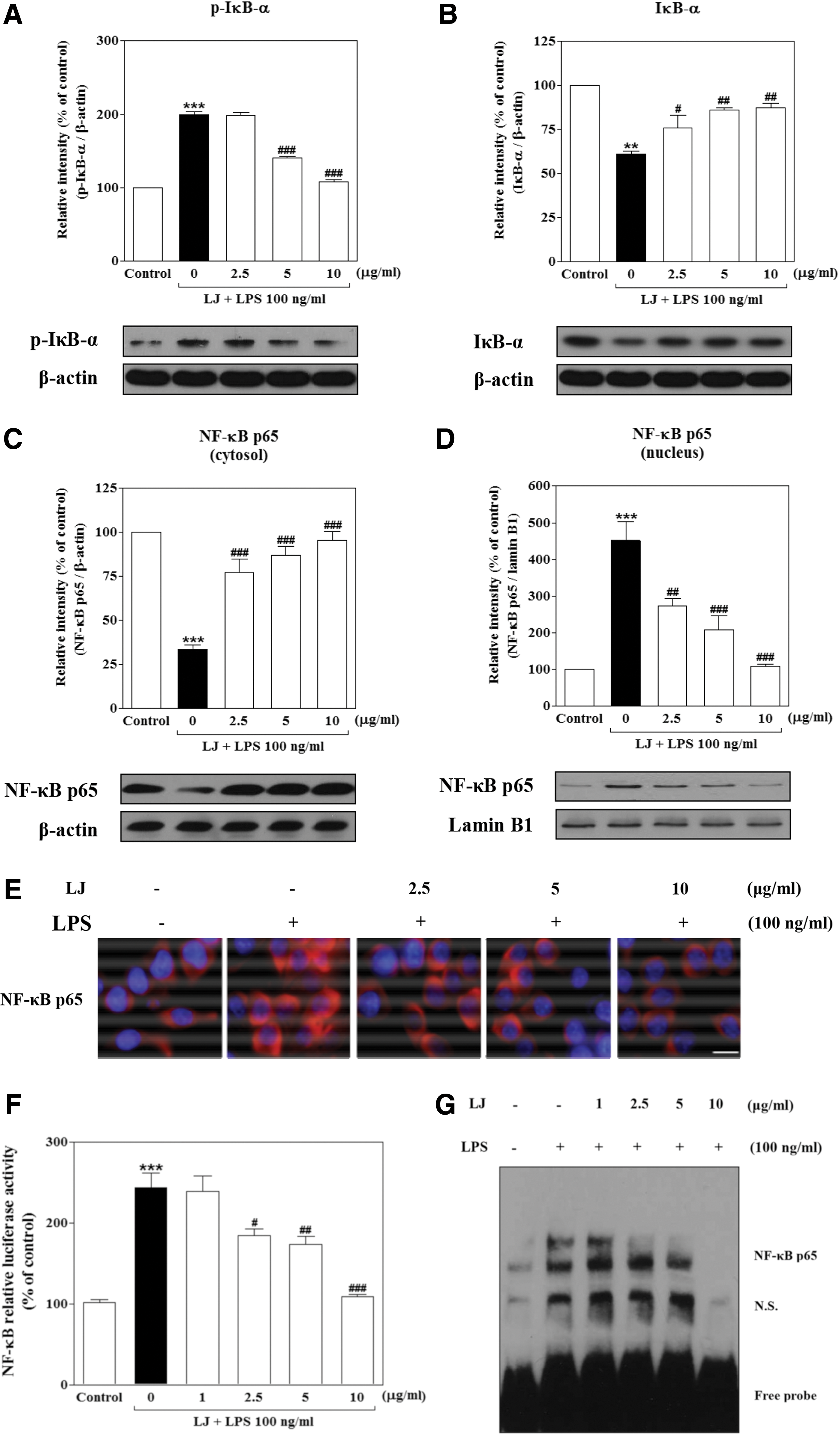
Proinflammatory responses and cytokine production are tightly regulated by signaling molecules such as IκB-α. We therefore used western blots to evaluate the cytoplasmic levels of this molecule under LPS-stimulating and LJ-inhibiting conditions. Treatment with LPS significantly increased the phosphorylation of p-IκB-α to 199.30%±4.57% of the control value (Fig. 6A, P<.001), whereas IκB-α significantly increased degradation to 60.97%±1.63% of the control value (Fig. 6B, P<.01). However, this increase in phosphorylation of p-IκB-α was significantly inhibited by 5 and 10 μg/mL of LJ to 140.30%±1.80% and 107.40%±3.58% of the control values, respectively (P<.001). On the other hand, pretreatment with 2.5, 5, and 10 μg/mL of LJ significantly inhibited degradation of IκB-α to 75.93%±7.10%, 85.97%±1.57%, and 87.37%±2.57% of control values, respectively (P<.05 and P<.01). Next, we investigated whether LJ inhibits localization of NF-κB from the cytosol to the nucleus using western blots and immunocytochemistry. Treatment with LPS significantly altered the cellular localization of NF-κB in cytosolic and nuclear fractions. Specifically, the levels of cytosolic and nuclear NF-κB in LPS-induced BV-2 microglial cells following LPS treatment were 33.31%±2.58% and 451.10%±51.81% of the control values, respectively (Fig. 6C, B, P<.01 and P<.001). However, NF-κB localization from the cytosol to the nucleus was significantly altered by 2.5 μg/mL LJ to 86%±1.54% and 148%±9.64% of the control values, respectively (P<.01 and P<.001). Pretreatment with 5 μg/mL of LJ also significantly altered NF-κB localization from the cytosol to the nucleus to 87.19%±4.98% and 207.90%±37.79% of the control values, respectively (P<.001), while 10 μg/mL of LJ inhibited NF-κB localization even more strongly to 95.35%±5.04% and 108.10%±6.63% of control values, respectively (P<.001). Immunocytochemistry analysis clearly confirmed the intracellular localization of NF-κB activation in LPS-induced cells (Fig. 6E). Specifically, treatment with LJ significantly blocked LPS-induced intracellular translocation of NF-κB from the cytosol to the nucleus in BV-2 microglial cells.
LJ inhibits both LPS-induced phosphorylation and degradation of IκB-α
Considering the inhibitory effects of LJ on LPS-induced activation of NF-κB localization, we measured the activation of NF-κB through NF-κB transcriptional activity by luciferase in BV-2 microglial cells transfected with an NF-κB reporter construct. As shown in Figure 6F, treatment with LPS significantly elevated the activation of NF-κB to 243.50%±18.85% of the control value (P<.001). However, activation of NF-κB was significantly inhibited by 2.5, 5, and 10 μg/mL of LJ to 184.30%±8.24%, 173.60%±10.30%, and 109.40%±2.11% of the control values, respectively (P<.05, P<.01, and P<.001). Finally, we asked whether LJ modulates the activation of NF-κB transcription factor binding to DNA following induction by LPS in BV-2 microglial cells by EMSA. Our results showed that treatment with LPS significantly increased the DNA-binding activity of NF-κB (Fig. 6G). In contrast, pretreatment with LJ significantly suppressed the increased DNA-binding activity of NF-κB induced by LPS.
Discussion
LJ has been shown to have anti-inflammatory properties in various experimental models. However, the specific effects of LJ on microglial cells have not been characterized. Therefore, in the present study, we examined the effects of LJ on LPS-stimulated inflammatory responses in BV-2 microglia cells. Specifically, we evaluated activation of inflammatory molecules and subsequent proinflammatory responses, as well as production of cytokines, chemokines, and ROS. To further understand the molecular mechanisms of LJ in LPS-treated BV-2 microglial cells, we investigated the inhibitory effects of LJ on LPS-stimulated phosphorylation of MAPKs, PI3K/Akt, and Jak1/STAT1/3, as well as on the activation of NF-κB p65.
NO and PGE2 are key inflammatory and neurotoxic mediators in inflammation and are responsible for injury to the CNS as well as several CNS diseases. 26 Indeed, many studies have demonstrated that abnormally high levels of NO and PGE2 are found in various types of brain injuries, and neurodegenerative diseases are caused by excessive expression of iNOS and COX-2 enzymes. 27,28 Thus, treatment with inhibitors of iNOS and COX-2 can facilitate neuroprotection against LPS-induced neurotoxicity, suggesting that NO and PGE2 are important mediators of neurotoxicity. 29 In the present study, we determined whether the inhibitory effects of LJ on LPS-stimulated NO and PGE2 production were related to induction of iNOS and COX-2. Interestingly, treatment with LJ significantly inhibited both NO and PGE2 production in a concentration-dependent manner and these inhibitory effects were mediated by downregulation of COX-2 and iNOS at the protein and mRNA levels. Together, these results suggested that LJ may be a promising candidate to inhibit the primary steps of inflammatory pathways.
TNF-α and IL-1β are the two main proinflammatory cytokines produced by activated microglia during CNS inflammation caused by the disruption of the blood–brain barrier. Excessive production of TNF-α and IL-1β has been linked to many neurodegenerative diseases, such as AD, PD, and HD. 30 –32 Overproduction of proinflammatory cytokines from activated microglial cells has a detrimental effect on neuronal cells. In addition, MCP-1 is a particularly important chemokine that is primarily responsible for the initiation and progression of proinflammatory responses by promoting migration and recruitment of inflammatory cells. 33,34 Thus, inhibition of cytokine and chemokine production or function serves as a key mechanism in the control of CNS inflammation. Accordingly, we investigated whether LJ inhibits LPS-induced production of proinflammatory cytokines in BV-2 microglial cells. Our data showed that LJ significantly inhibited LPS-induced expression of mRNA levels of TNF-α, IL-1β, and MCP-1 mRNA and protein secretion. These results suggested that in LPS-activated BV-2 microglial cells, LJ inhibits the production of proinflammatory cytokines and chemokines at the level of transcription.
MMP-9 plays a fundamental role in normal physiological processes and also contributes to several pathologies associated with uncontrolled tissue degradation. In addition, several recent reports have indicated that MMP-9 is secreted from activated microglia and is involved in neuronal damage and subsequent neuroinflammatory processes in AD, PD, HD, and stroke. 35 Thus, controlling microglia-mediated MMP-9 production has been suggested as a possible therapeutic approach to treat CNS inflammation. 5,36 Several recent reports have demonstrated that natural products can inhibit MMP-9 and are thus considered to be potential therapeutic agents for alleviating neurodegenerative diseases. 37,38 In the present study, we investigated the effect of LJ on production of MMP-9 in LPS-stimulated BV-2 microglial cells. RT-PCR analysis revealed that LJ significantly inhibited the expression of MMP-9 mRNA levels in LPS-stimulated BV-2 microglial cells. Consistent with this result, treatment with LJ strongly attenuated LPS-induced MMP-9 secretion in BV-2 microglial cells. Taken together, our data suggest that LJ can block proinflammatory responses in activated BV-2 microglial cells with activated MMP-9 production and amplified inflammatory cascade signaling.
Excessive ROS generation by microglia in the brain contributes to neuronal damage involved in neurodegenerative diseases. 39,40 In addition, ROS amplify inflammatory signals during chronic inflammation in microglia through activation of kinases, such as JNK, p38 MAPKs, and PI3K/Akt, leading to the activation of NF-κB and subsequent induction of overexpression of inflammatory molecules in activated microglial cells. 38,41 –43 To evaluate whether the inhibitory effects of LJ on intracellular ROS accumulation are related to upstream activation of NF-κB and phosphorylation of MAPKs and PI3K/Akt, we examined intracellular ROS production in BV-2 microglial cells. Our results indicated that LJ significantly inhibited LPS-induced intracellular ROS production in BV-2 microglial cells, suggesting a possible mechanism for the inhibitory effects of LJ on both activation of NF-κB and phosphorylation of MAPKs and PI3K/Akt. Furthermore, potential inhibition of ROS generation by LJ could lead to the inhibition of signaling pathway-dependent production of proinflammatory mediators and/or cytokines, thereby suppressing inflammation.
It has been shown that intracellular signaling pathway molecules, such as MAPKs and PI3K/Akt, are activated by LPS and are involved in the regulation of inflammatory responses, cytokines, and chemokines in microglia cells. 44 –46 Recent studies have also shown that LPS stimulates NF-κB activation by both phosphorylating and degrading IκB following activation of IKK and phosphorylation of MAPKs and PI3K/Akt. 47 –49 Moreover, inhibition of the phosphorylation of MAPKs and PI3K/Akt also contributes to the inhibition of iNOS and COX-2 expression as well as inhibition of gene expression of cytokines and chemokines in microglial cells. 9,38,44,50 Although a previous report on the effects of LJ on NF-κB-mediated oxidative stress pathways received considerable attention, the mechanisms underlying the interactions of LJ with these signaling pathways are not fully understood. 20,21 In particular, it is not known whether LJ inhibits the phosphorylation of MAPKs and PI3K/Akt in microglial cells. Therefore, new experiments are needed to determine whether LJ tightly regulates the activation of MAPKs and PI3K/Akt in LPS-activated BV-2 microglial cells. In the present study, we found that LJ acted as a potent inhibitor of MAPK and PI3K/Akt phosphorylation. Together, these findings suggested that LJ is capable of disrupting key signal transduction pathways activated by LPS in BV-2 microglial cells that subsequently prevent production of neuroinflammatory molecules.
JAK/STAT inflammatory signaling has recently been reported to play a role in inflammatory responses in the brain. 51 The JAK/STAT signaling pathway has also been reported to mediate actions of growth factors, hormones, and cytokines. 52 In addition to activation of NF-κB and ROS production signaling in LPS-induced microglial cells, LPS also activates the JAK/STAT signaling pathway, which itself is important for chemokine expression. 53,54 However, the mechanisms underlying interactions of LJ with these signaling pathways are poorly understood. Thus, we tested whether the anti-inflammatory effects of LJ were related to the suppression of JAK/STAT activation in BV-2 microglial cells. Specifically, we showed that LJ suppressed LPS-induced phosphorylation of JAK1/STAT1/3. In addition, the inhibitory action of LJ on phosphorylation of JAK1 correlated with its effects on phosphorylation of STAT1/3. Therefore, phosphorylation of STATs depends on the activation of JAKs. Collectively, our results suggest that LJ inhibits LPS-induced initiation of the JAK/STATs signaling cascade at least at the level of production of cytokines, chemokines, and ROS, as well as NF-κB activation.
NF-κB is known as a pleiotropic regulator of various genes involved in the production of numerous proinflammatory cytokines and enzymes related to the inflammatory process. Indeed, NF-κB is one of the central regulators of microglial responses with respect to activating stimuli, including LPS and cytokines. 55 In terms of the mechanism, NF-κB is inactive when bound to IκB-α in the cytosol. However, in response to stress, phosphorylated IκB-α is degraded through selective ubiquitination, resulting in the activation of NF-κB. Activated NF-κB then localizes to the nucleus and binds to the promoter regions of proinflammatory molecules, thereby upregulating gene expression. 56 The unregulated prolonged production of these molecules from activated microglial cells leads to CNS cell damage and death, especially neurons. 57 Since NF-κB is known to play a critical role in the inflammatory response, we also investigated whether LJ inhibits the activation of NF-κB translocation and examined the effect of LJ on the DNA-binding activity of NF-κB with EMSA. Our results showed that LJ prevents LPS-induced activation of NF-κB localization and consequently reduces LPS-induced DNA–protein-binding activity of NF-κB. Therefore, inhibition by LJ of NF-κB signaling pathways in BV-2 microglial cells might lead to downregulation of proinflammatory mediators, thereby resulting in an anti-inflammatory effect.
The anti-inflammatory effects of LJ are likely due to specific compounds present in the extract. The major active constituents that have been identified in LJ comprise iridoid glycosides and flavonoids, such as sweroside, loganin, hyperoside, chlorogenic acid (CGA), luteolin, and caffeic acid. 58,59 Recently, we reported that loganin and CGA ameliorate scopolamine-induced learning and memory deficits in mice through antiacetylcholinesterase and/or antioxidative stress activities. 60,61 In addition, luteolin has preventive effects on Aβ 1–40-induced cognitive impairments as measured by behavioral performance tests such as passive avoidance and Morris water maze tests. 62 Although the presence of LJ in the brain was not determined, evidence suggests that flavonoids in LJ, including CGA and luteolin, are generally able to penetrate the blood–brain barrier and can be detected in the brain. 63,64 Thus, it is reasonable to predict that circulating LJ had access to the microglial cell compartment.
LJ could conceivably reduce immune responses in the brain through undefined peripheral mechanisms, which would also manifest as reduced brain inflammation. However, the primary components of LJ have been reported to be responsible for various pharmacological actions, particularly CGA, and were recently demonstrated to have anti-inflammatory effects on LPS-induced proinflammatory responses through NF-κB activation in RAW 264.7 cells. 65 In our study, high-performance liquid chromatography (HPLC) analysis of the LJ extract used in this study identified CGA as a peak compared with an LJ standard sample (Supplementary Fig. S2). CGA might act independently or synergistically by regulating multiple signaling pathways in blocking neuroinflammation; however, the actual concentrations of these potential compounds in LJ extract that are able to reach the brain and produce beneficial effects remain in question and will require further research.
In summary, the results presented in this study identify a potential anti-inflammatory effect of LJ in BV-2 microglial cells. In LPS-stimulated BV-2 microglial cells, LJ significantly inhibited the production of the inflammatory mediators, NO and PGE2, and also suppressed expression and release of molecules involved in inflammation, including iNOS, COX-2, TNF-α, IL-1β, MCP-1, and MMP-9. These inhibitory effects were associated with the activity of LJ in suppressing the phosphorylation of MAPKs, PI3K/Akt, and Jak1/STAT1/3 and activating NF-κB. The proposed signaling mechanisms involved in the antineuroinflammatory effects of LJ are shown in Supplementary Figure S3. Considering the critical roles of the various inflammatory mediators of inflammation, our results suggest that LJ has the potential to be a therapeutic agent for the treatment of inflammation in neurodegenerative diseases such as AD and PD.
The neuroprotective effects of LJ should be further investigated in in vivo models to provide definitive evidence for its potential role as a therapeutic agent for neurodegenerative diseases such as AD and PD. Although the present study did not evaluate whether LJ inhibits inflammation-related neuronal damage in vivo, we demonstrated that LJ exerts anti-inflammatory activity in BV-2 microglial cells. To confirm the involvement of multiple signaling pathways in the mechanism mediating the neuroprotective effects of LJ against neurotoxicity, we are currently working to better understand the role of LJ in neurodegenerative diseases such as AD and PD. These studies will require extensive evaluations regarding the neuroprotective effects of LJ in animal models, including those where neurotoxins such as Aβ and MPTP are regionally injected into the brain.
Footnotes
Acknowledgment
This research was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF-2012R1A5A2A28671860 and NRF-2011-00503) funded by the Ministry of Education, Science and Technology, Republic of Korea.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.