
Abstract
The purposes of this study were to examine the characteristics of Helicobacter pylori and the effect of Rumex Aquaticus Herba extract on the expression of cytokines in H. pylori-infected gastric epithelial cells. Cultured human adenocarcinoma gastric cells (AGS) were infected by H. pylori in RPMI 1640 media. Cell growth was measured by trypan blue assay. Western blot analysis was performed to investigate effect of extract containing Quercetin-3-O-β-
Introduction
H
Rumex Aquaticus is a member of the Polygonaceae family, and this plant has been used to treat infestation, diarrhea, edema, jaundice, and constipation in traditional oriental medicine. Quercetin-3-O-β-
Materials and Methods
Reagents
ECQ was kindly provided by Prof. W.K. Whang (Pharmacal Botany Resources Laboratory, Chung-Ang University College of Pharmacy, Seoul, Korea). ECQ was extracted from Rumex Aquaticus with ethanol containing 10.7% QGC, of which the chemical structure was shown in previous study. 15 Bovine serum albumin, ethylene glycol-bis-(β-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA), ethylenediamine tetra acetic acid (EDTA), trypan blue solution (0.4%), leupeptin, aprotinin, β-mercaptoethanol, phenylmethylsulfonylfluoride (PMSF), triton X-100, sodium dodecyl sulfate (SDS), hank's balanced salt solution-modified (HBSS), clarithromycin, omeprazole, amoxicillin, christensen's urea agar, and urea were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Fetal bovine serum (FBS), antibiotic-antimycotic (penicillin, streptomycin, and amphotericin B), and trypsin-EDTA were purchased from Invitrogen (Grand Island, NY, USA). RPMI 1640, tris-buffered saline, and phosphate-buffered saline (PBS) were purchased from Welgene, Inc. (Daegu, Korea). Anti-IκB-α, anti-IL-8, and anti-IL-1β antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); goat anti-rabbit IgG-HRP, goat anti-mouse IgG-HRP, and rabbit anti-goat IgG-HRP from Zymed Laboratories, Inc. (Eccles Avenue, CA, USA). The rainbow prestained molecular weight marker was purchased from Elpis Biotechnology (Daejeon, Korea). Enhanced chemiluminescence agents were purchased from PerkinElmer Life Sciences (Boston, MA, USA). Ammonium persulfate, N,N,N’,N’-tetramethylethylendiamine (TEMED), nitrocellulose membrane, tris/glycine/SDS buffer, tris/glycine buffer, restore TM western blot stripping buffer, and 30% acrylamide/bis solution were purchased from BioRad (Richmond, CA, USA).
Bacterial strain
An H. pylori strain (HP99) was isolated from gastric antral mucosa obtained from a Korean patient with a duodenal ulcer and was identified as CagA+ and VacA+. 16 HP99 was kindly supplied by Dr. J.G. Kim (Chung Ang University College of Medicine, Seoul, Korea). Bacteria were inoculated onto blood agar plates (Hanil Komed, Songnam, Korea) at 37°C under microaerophilic conditions using a 10% CO2 incubator (Vision Scientific, Bucheon, Korea).
Cell culture and H. pylori stimulation
The human gastric epithelial cell line AGS (gastric adenocarcinoma, KCLB 21739) was obtained from the Korean Cell Line Bank (Seoul, Korea). Cells were grown in complete medium, consisting of RPMI 1640 medium supplemented with 10% FBS, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 0.25 μg/mL amphotericin B. AGS cells were seeded and cultured to reach 80% confluency. H. pylori was harvested, washed with PBS, and resuspended into antibiotic-free cell culture medium. H. pylori was added to AGS cells at a bacterium/cells ratio of 100:1, and cultured for the indicated time periods.
Measurements of cell proliferation
The cell proliferation was determined by Trypan blue exclusion assay. The cells were plated at a density of 1 × 105 cells/well in six-well plates and maintained in RPMI 1640 containing 10% FBS at 37°C under microaerophilic conditions for 3 days. The media was changed with serum-free RPMI and incubated for 24 h to arrest cell growth and silence gene activity, followed by treatment with H. pylori at a bacterium/cells ratio of 100:1 for the indicated time periods. After incubation, the cells were washed twice with HBSS and harvested. The number of viable cells was determined using light microscopy and counting cells that excluded the 0.2% trypan blue dye. Cells were counted in a randomized manner using a hematocytometer.
Urease activity assay
Urease activity was measured to test H. pylori's ability to secrete the urease enzyme, which catalyzes the conversion of urea to ammonia and carbon dioxide. Briefly, autoclaved christensen's urea agar was mixed with urea and a bacterial suspension containing the liquid reaction mixture was added to the tube, followed by incubation for 5 h at 37°C under microaerophilic conditions. The reaction was observed for changes in the color, which would indicate an increase in the pH due to the production of ammonia.
Western blot analysis
AGS cells were allowed to grow for 3 days, and were stimulated as described. Cell lysates were prepared using lysis buffer (20 mM tris-HCl [pH7.4], 0.5 mM EDTA, 0.5 mM EGTA, 10 mg/mL triton X-100, 100 μg/mL SDS, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM PMSF, and 0.7 μg/mL β-mercaptoethanol). The lysates were subjected to electrophoresis on a 12% SDS-PAGE, transferred to nitrocellulose membranes using the Power Pac 1000 (Bio-Rad, Melville, NY, USA) power supply and examined for expression of IκB-α, IL-8, and IL-1β. Actin was used as a loading control. The results were analyzed by Quantity One analysis software (Bio-Rad Chemical Division, Richmond, CA, USA).
Data analysis
All data are expressed as mean ± SEM of 3–4 experiments. Statistical differences among the groups were analyzed by Student's t-test. Data were considered at P < .05.
Results
Changes in cell growth rate during H. pylori infection in AGS cells
Trypan blue assays were performed in cultured AGS cells to investigate the cytotoxic effects of H. pylori. As shown in Figure 1, serum-starved AGS cells were incubated with H. pylori at a multiplicity of infection of 100 for 0, 3, 6, 12, and 24 h, and the cell proliferation was measured using the trypan blue assay. Apoptotic cells were more apparent in cells incubated with H. pylori than those in the control groups at every time point, with the most significant difference seen at 24 h. Infected cell numbers were gradually increased time-dependently. However, the number of live cells in the H. pylori group was decreased to ∼72% compared with cells in the control group.

Effect of Helicobacter pylori on the cell proliferation of AGS cells. AGS cells were incubated with H. pylori (multiplicity of infection [MOI]: 100) for the indicated time periods. Cell proliferation was estimated using Trypan blue assay. Data are expressed as mean ± standard error (SE) of three experiments (Student's t-test; *P < .05 vs. control, **P < .01 vs. control). AGS, adenocarcinoma gastric cell line.
Expressions of IκB-α, IL-8, and IL-1β after H. pylori incubation
To investigate when the proinflammatory proteins induced by H. pylori would reach maximal expression levels in AGS cells, serum-starved AGS cells were exposed to H. pylori for 0, 0.5, 1, 2, 4, 6, 8, and 12 h and IκB-α, IL-8, and IL-1β protein expressions were detected by western blot analysis. H. pylori infection significantly degraded IκB-α, which translated into the activation of NF-κB (Fig. 2a). IκB-α expression levels increased after 60 min. It has been reported that H. pylori infection increases the expression of some proinflammatory cytokines, including IL-8 and IL-1β, in gastric mucosa. 17 After incubation with H. pylori, an increase in IL-8 levels was observed relative to cells incubated with medium as a control. IL-8 levels rose steadily and peaked at 8 h, and slightly declined by 12 h (Fig. 2b). AGS cells incubated with H. pylori for 4 h resulted in time-dependent increase in IL-1β protein. After 4 h, the IL-1β expression level decreased (Fig. 2c). Based on these results, experiments investigating IκB-α in AGS cells were performed with H. pylori infection for 1 h and experiments looking at IL-8 and IL-1β were conducted for 8 and 4 h, respectively.

H. pylori-induced
H. pylori-induced IL-1β and IL-8 expressions are decreased by ECQ pretreatment
To examine whether ECQ attenuates IκB-α, IL-8, and IL-1β expressions in AGS cells, the cells were pretreated with ECQ for 12 h at 12.5, 25, 50, and 75 μg/mL and exposed to H. pylori. IκB-α, IL-8, and IL-1β expressions were measured by western blot analysis. There was no significant reduction in H. pylori-induced IκB-α degradation in cells pretreated with ECQ (Fig. 3a). ECQ suppressed IL-8 production by H. pylori in a dose-dependent manner (Fig. 3b). Moreover, IL-1β levels were also significantly downregulated by ECQ suggesting it potently inhibited H. pylori-induced inflammation in AGS gastric epithelial cells (Fig. 3c).

Effect of ECQ on
To further address ECQ's ability to inhibit H. pylori-induced inflammation, we examined the effects of ECQ with various combinations of drugs commonly used to treat H. pylori infection (Fig. 4). ECQ in combination with amoxicillin and clarithromycin reduced cytokine expressions in AGS cells more effectively than the ECQ treatment and amoxicillin plus clarithromycin treatment groups. However, omeprazole treatment was not efficacious in the prevention of H. pylori-induced inflammation.
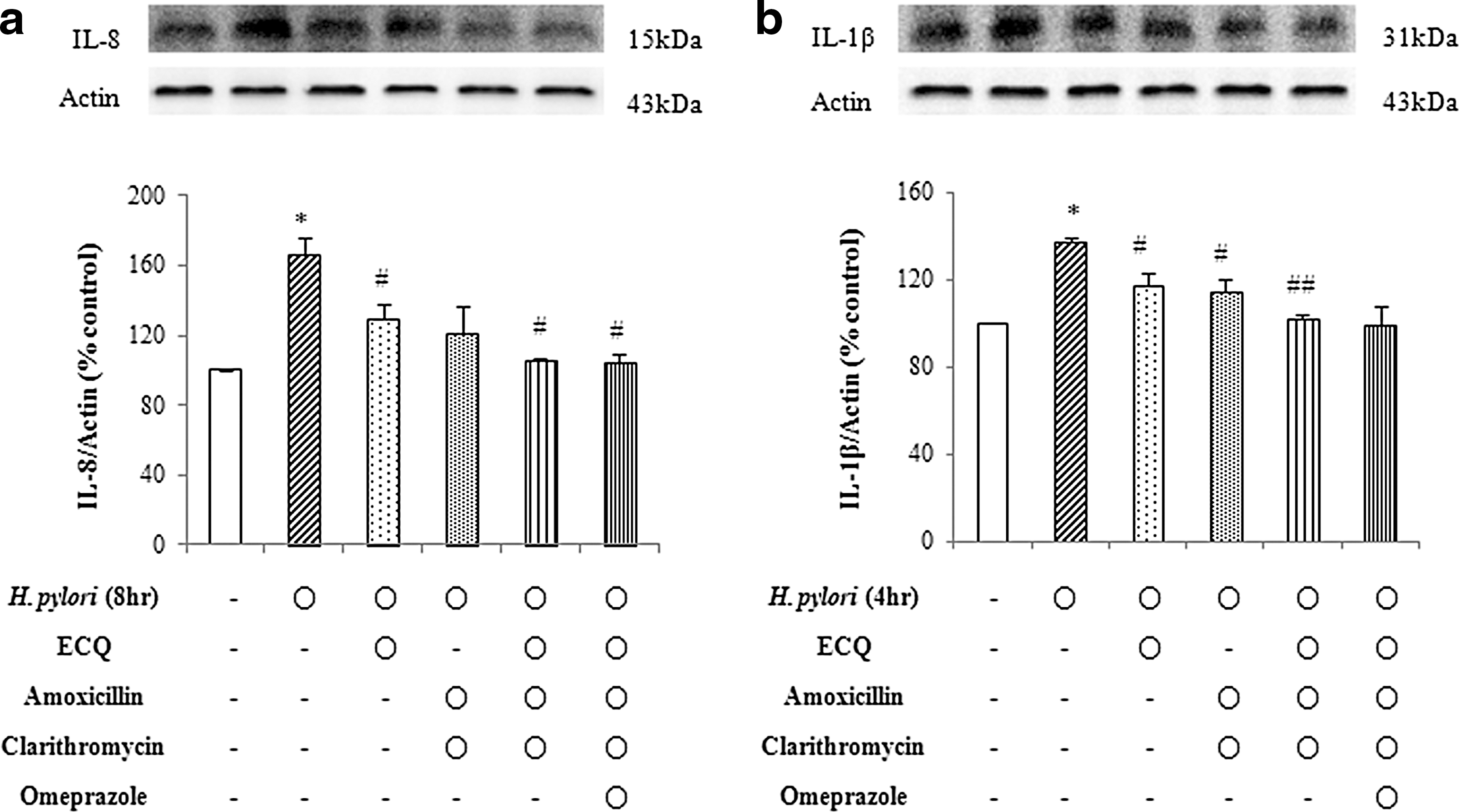
Effect of ECQ, amoxicillin, clarithromycin, and omeprazole on
Urease activity of H. pylori is not altered by treating with ECQ
To investigate whether ECQ inhibits the urease activity of H. pylori, an assay using Christensen's Agar Base with urea was conducted. First, urease activity test was performed with or without yeast extract. Without yeast extract, the urease activity of H. pylori was lower compared with the activity in the presence of yeast (Data not shown). We also examined the urease activity of H. pylori incubated with various concentration of ECQ to determine whether it inhibited the urease activity of H. pylori (Fig. 5). H. pylori was combined with Christensen's Agar Base and ECQ. After 5 h, few changes in the liquid medium's pH due to the production of ammonia by urease detected via phenol red, indicating that ECQ did not inhibit the urease activity of H. pylori.

Effect of ECQ on urease activity of H. pylori in AGS cells. Cultured H. pylori was incubated in combination with Christensen's Agar Base with urea and ECQ at the indicated concentration. All the reactions were carried out at 37°C. When the urease in H. pylori increases the pH of the medium, the phenol red causes it to change from yellow to red. Color images available online at
Discussion
The main findings of this study are that H. pylori mediates the degradation of IκB-α and induces proinflammatory cytokines in AGS gastric epithelial cells. In addition, ECQ could be used as a chemoprotective agent for gastritis or peptic ulcers induced by H. pylori. Inflammation mediated by IL-8 and IL-1β is a central theme in H. pylori-induced gastritis and ulcers. Some reports have demonstrated that IL-1β plays an important role in synthesizing IL-8 in response to the inflammatory signals in gastric epithelial cells. 18,19 IL-1β also upregulates many proinflammatory proteins, including IL-8, in other cells types such as esophageal and colon epithelial cells. 20,21 IL-8 is a chemokine that attracts neutrophils and eosinophils. 22 These white blood cells are implicated in gastritis and GI tract inflammation. Therefore, IL-8 might be a potential target for therapeutic intervention in gastric inflammation.
Numerous medicinal products have been used for gastritis and ulcers, regardless of their side effects. Many plant extracts are displaying efficacy in treating gastritis, and have become one of the most promising sources of new drugs. 23 Flavonoids such as QCG have anti-inflammatory, antioxidant, antiallergic, anticarcinogenic, and antiviral activities against various diseases. 24
Quercetin has been identified to have anti-inflammatory, 25 antithrombotic, 26 antitumor, 27 and antibacterial 28 effects. QGC is a quercetin derivative, which is extracted from Rumex Aquaticus Herba. In our previous study, QGC inhibited gastric acid output, which prevented the development of reflux esophagitis. 13 QGC also reduced ROS generation and ERK activation induced by ethanol in cultured feline esophageal epithelial cells. 29 In addition, QGC prevented indomethacin-induced gastric mucosal injury by increasing mucus secretion and attenuating the release of free radical and proinflammatory cytokines. 30
In this study, cell growth inhibition induced by H. pylori increased time-dependently with apoptotic cell death. AGS gastric cell proliferation was slightly decreased in the H. pylori-infected groups compared with the control group over 24 h. These results indicate that H. pylori infection may continuously suppress the growth of AGS gastric epithelial cells.
Previous studies suggested that NF-κB activation might be a necessary precondition for IL-8 upregulation in response to H. pylori. 31 –33 H. pylori adheres to gastric epithelial cells and activates NF-κB, a known regulator of IL-8 production. 31,34 –36 Activation of NF-κB is directly mediated by the degradation of IκB. In this study, IκB-α degradation was evident within 30 min of H. pylori infection and peaked at 1 h.
Our result shows that cytokine expression gradually increased in H. pylori-infected AGS cells, indicating that H. pylori-induced inflammation may mediate an upregulation of IL-8 and IL-1β through the degradation of IκB-α. In addition, we found that pretreatment with ECQ markedly decreased the expressions of H. pylori-induced cytokines. The initial migration and activation of inflammatory cells is known to rely on various proinflammatory cytokines production, especially IL-8. 37 These results indicate that the protective effect of ECQ might be related to anti-inflammatory properties that prevent cytokine formation.
Our results also suggested a possible application of ECQ in combination with commonly used antibiotics. In addition to ECQ, two antibiotics displayed an inhibitory effect against H. pylori-induced inflammation in AGS cells. When combined with clarithromycin and amoxicillin, ECQ demonstrated cumulative activity against H. pylori-induced proinflammatory cytokine expressions to almost basal levels. Our results are supported by in vivo studies that demonstrate the protective effects of ECQ on gastric mucosa in gastritis-induced rats. 38,39 The present results indicate that ECQ might have a useful role as part of a combination treatment with anti-H. pylori drugs in the treatment of diseases caused by the bacteria.
In this study, urease activity was detected in live H. pylori. Urease activity of H. pylori is important for gastric colonization and maintaining the cytoplasmic pH. 40,41 We also co-incubated ECQ and H. pylori in Christensen's Agar Base containing urea. However, H. pylori was able to change the pH in the presence of ECQ, indicating that the inhibitory mechanism of ECQ on H. pylori does not involve suppressing urease activity.
Taken together, our findings demonstrate that ECQ has its ability to inhibit the cellular processes critically involved in the pathogenesis of H. pylori, which induces the expressions of various inflammatory cytokines, such as IL-8 and IL-1β. Among the previously mentioned cytokines, the upregulation of IL-8 plays a pivotal role in the H. pylori-mediated signaling pathway in gastric epithelial cells. Further studies need to be conducted to obtain more detailed information about the molecular mechanism of ECQ, but this study supports its potential as a possible candidate for the treatment of gastric inflammation associated with H. pylori infection.
Footnotes
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (no. 2011-0012139).
Author Disclosure Statement
No competing financial interests exist.