
Abstract
Aggregation and deposition of beta-amyloid peptides (Aβ), a pathological hallmark of Alzheimer's disease, has been recognized as a potent activator of neuroinflammation and neuronal dysfunction. In this study, the underlying molecular mechanisms responsible for the neuroprotective effects of corilagin against Aβ 25–35-triggered neurotoxicity and inflammatory responses were investigated in PC12 cells. Pretreatment with corilagin effectively protected PC12 cells against Aβ 25–35-induced damage and apoptosis. Aβ 25–35 induced damage in PC12 cells as revealed by increased production of reactive oxygen species, caspase-3 activity, and cell cycle arrest was attenuated by corilagin pretreatment. Corilagin not only significantly suppressed the production of neurotoxic inflammatory mediators such as tumor necrosis factor-α, nitric oxide, and prostaglandin E2 but also downregulated cyclooxygenase-2 and inducible nitric oxide synthase expression in PC12 cells. It also exerted a beneficial effect by suppressing the degradation of inhibitor of κB (IκB)-α and subsequent activation of transcription factor nuclear factor κB (NF-κB), mostly through inhibition of extracellular signal-regulated kinase activity in comparison to c-Jun N-terminal kinase and p38 MAP kinase (p38) mitogen-activated protein kinase activity. These findings suggest that attenuation of Aβ 25–35-induced inflammatory responses by downregulating the NF-κB signaling pathway might be a valuable strategy for both Alzheimer's disease prevention and/or treatment.
Introduction
A
Nuclear factor κB (NF-κB) is a transcription factor that plays a vital role in the gene regulation associated with inflammation. It is activated in the neurons with Aβ deposition in the early stages of AD. 5 Binding of inhibitor of κB (IκB) to NF-κB, mostly in the form of heterodimer RelA(p65)/p50, causes NF-κB to be sequestered in the cytoplasm in the inactive form. When NF-κB-activating stimuli trigger the IκB kinase (IKK) complex, IKK phosphorylates IκB, and NF-κB is released into the nucleus, where it interacts with NF-κB binding elements in the promoter region of the target genes to modulate gene expression. 6 The translocation of NF-κB by Aβ induces transcription of proinflammatory mediators such as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), which then activate the circulating inflammatory mediators. 7,8 Furthermore, the NFκB signaling pathway is intimately linked to the activation of mitogen-activated protein kinases (MAPKs), which stimulate downstream transcription factors that promote inflammatory gene expression. 9 These signaling pathways are well characterized in mammals and include c-Jun N-terminal kinase (JNK), p38 MAP kinase (p38), and mitogenic signaling extracellular signal-regulated kinase 1/2 (ERK).
Corilagin [β-1-O-galloyl-3,6-(R)-hexahydroxydiphenoyl-D-glucose], a member of the ellagitannin family, has been discovered in several species of Geranium (Geraniaceae) and Phyllanthus (Phyllanthaceae). 10 Although a wide spectrum of pharmacological properties, including antioxidative, antihypertensive, antitumor, and antiatherogenic effects, 11 –13 have been reported, little research has been devoted to investigating the inhibitory activities of corilagin against Aβ-stimulated neuroinflammatory toxicity.
In our previous study, corilagin isolated from the EtOAc fraction of Geranium thunbergii not only inhibited the recombinant human β-secretase (BACE1) activity but also noncompetitively bound with and suppressed BACE1 in an in silico docking model system. 14 To extend our knowledge of the neuroprotective effects of corilagin, its anti-inflammatory properties along with the possible underlying mechanisms (particularly focused on NF-κB and MAPK signaling pathway) were investigated for the first time in Aβ 25–35-treated PC12 cells.
Materials and Methods
Materials
Corilagin (Fig. 1A) was isolated (>99% purity) and its chemical structure was determined in our previous study. 14 Aβ 25–35, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), resveratrol (used as a positive control), and Griess reagent were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Prostaglandin E2 (PGE2) immunoassay kit was purchased from R&D (Minneapolis, MN, USA). RPMI 1640 medium, phosphate-buffered saline (PBS), donor equine serum, trypsin 0.25% solution, and penicillin were supplied by Hyclone Laboratories (Logan, UT, USA). Fetal bovine serum (FBS) was obtained from PAA Laboratories (Linz, Austria). Hank's balanced salt solution (HBSS), N2 supplement, and RPMI 1640 phenol red-free medium were obtained from Gibco BRL (Grand Island, NY, USA). CM-2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and Hoechst 33342 dye were obtained from Molecular Probes (Eugene, OR, USA), and caspase-3/CPP32 colorimetric assay kit was purchased from BioVision (Pato Alto, CA, USA). The specific antibodies for tumor necrosis factor (TNF)-α, iNOS, COX-2, β-actin, monoclonal antibodies, and peroxidase-conjugated secondary antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-phospho-p65, phospho-IκB-α, phospho-JNK, phospho-ERK, and phospho-p38 monoclonal antibodies were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Muse Count & Viability Kit and Muse Annexin V & Dead Cell Kit were purchased from Merck Millipore (Billerica, MA, USA). All other chemicals and regents were purchased from Sigma-Aldrich Chemical Co.
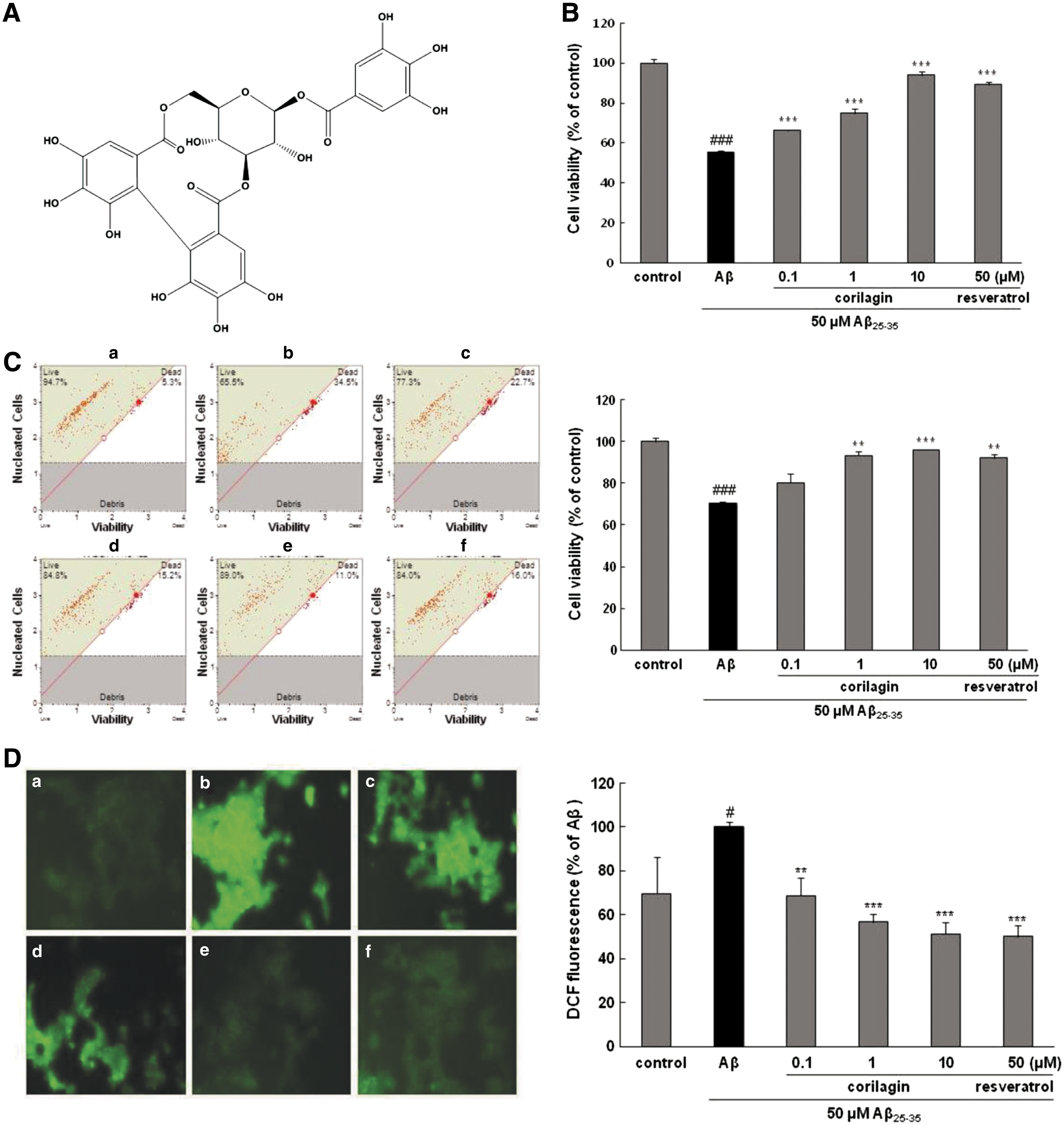
Effects of corilagin on Aβ
25–35-induced PC12 cell death.
Cell culture and Aβ preparation
PC12 cells were grown in the RPMI 1640 medium supplemented with 10% heat-inactivated horse serum and 5% FBS at 37°C in a humidified 95% air/5% CO2 incubator. Aβ 25–35 was dissolved in dimethyl sulfoxide at an initial concentration of 10 mM and was diluted with PBS. Aβ 25–35 solution was then incubated at 37°C for 48 h to aggregate before use.
Cell viability assay
Cells were plated at the density of 1 × 105 cells/well in a 96-well plate. After incubation with corilagin for 1 h, the cells were treated with 50 μM Aβ 25–35 for 24 h. Thereafter, 0.5 mg/mL of MTT reagent (final concentration) was added to the incubated cells and then formazan reduction product was determined using a microplate reader at a wavelength of 570 nm (ELX808; Biotek, Winooski, VT, USA).
Measurement of intracellular reactive oxygen species accumulation
Cells were incubated at 37°C with 50 μM Aβ 25–35 for 24 h with or without pretreatment with corilagin. Then, the cells were further incubated in 10 μM CM-H2DCFDA for an additional 30 min in the dark. After replacing the medium with 100 μL HBSS, reactive oxygen species (ROS) generation was determined using a fluorescence spectrophotometer (FLX800; Biotek) at 485 ex/528 em.
Determination of apoptotic cells
PC12 cells cultured on coverslips were treated with corilagin for 1 h before the addition of 50 μM of Aβ 25–35 and were then incubated for another 24 h. After incubation, the cells were fixed in 4% formaldehyde in PBS and stained with 1 μg/mL Hoechst 33342 solution. Hoechst-stained cells were mounted on slide glasses and examined under a fluorescence microscope ( × 400; Olympus, Tokyo, Japan).
Assay for caspase-3 activity
Cells were lysed for 10 min in an ice bath, and the lysates were centrifuged at 9,800 g for 1 min at 4°C. The supernatant was incubated with substrate peptide in 50 μL of incubation buffer at 37°C for 1 h. Protein concentration was confirmed with a BCA assay kit (Pierce Biotechnology, Rockford, IL, USA). Protein samples (50 μg) were incubated with DEVD-pNA in reaction buffer at 37°C for 1 h. The production of pNA was monitored in a plate reader at 405 nm (ELX808; Biotek).
Measurement of NO and PGE2 production
Briefly, cells were pretreated with corilagin for 1 h and then incubated with Aβ 25–35. After 24 h, the supernatants were collected and mixed with the same amount of Griess reagent. Samples were incubated at room temperature for 15 min, and the absorbance was subsequently read at 540 nm using a microplate reader (ELX808; Biotek).
PC12 cells were preincubated with corilagin in the presence of Aβ 25–35 for 24 h. Following the manufacturer's instructions, the culture medium supernatant was collected for determination of PGE2 concentration by the enzyme-linked immunosorbent assay (ELISA) kit. The changes in absorbance at 405 nm were measured using a spectrophotometric microplate reader (ELX 808; Biotek).
Flow cytometry analysis
Cell viability, apoptotic cell number, and number of cell cycle were determined using the Muse™ count & viability kit, annexin V and dead cell kit, and cell cycle kit, respectively, according to the manufacturer's instructions. Cell suspensions were added to Muse cell reagent and analyzed using the Muse cell analyzer (Merck Millipore).
Western blot analysis
The whole cell lysates were centrifuged at 16,500 g for 10 min at 4°C and protein concentrations were analyzed by the BCA method. Equal amounts of proteins (40 μg) were separated by 10% sodium dodecyl sulfate-acrylamide gel at 130 V and transblotted onto polyvinylidene difluoride (PVDF) membranes (Merck Millipore). Blots were blocked for 2 h at room temperature with 5% nonfat dried milk in the PBST buffer (0.1% Tween 20 in PBS), and membranes were incubated overnight with specific antibodies at 4°C. The following antibodies were used to probe the membranes: TNF-α (1:1000), iNOS (1:500), COX-2 (1:1000), phospho-p65 (1:1000), phosphor-IκB-α (1:800), phospho-ERK 1/2 (1:1000), phospho-JNK (1:500), and phospho-p38 (1:500). After washing with PBST, membranes were probed with secondary antibody (horseradish peroxidase conjugated rabbit or goat polyclonal IgG) at room temperature for 1 h. The relative density of the protein bands was quantified using the AE-9160 EZ-capture chemiluminescence imaging system (ATTO, Tokyo, Japan) with the EzWestLumi plus chemiluminescent detection reagent (ATTO).
Statistics
All data in the text and figures are expressed as means ± standard deviation values. The values were compared with the control using analysis of variance followed by unpaired Student's t-test. P values of *P < .05, **P < .01, and ***P < .001 were regarded as statistically significant. All data points represent the mean of triplicates.
Results
Corilagin protected Aβ 25–35-induced neuronal cell death and ROS accumulation
Treating PC12 cells with Aβ 25–35 decreased cell viability to 56.77% ± 1.99% in comparison to that of untreated control cells (100%) (P < .001, Fig. 1B). However, pretreatment with corilagin before exposure to Aβ 25–35 increased the cell viability in a dose-dependent manner by 68.67% ± 4.56%, 80.78% ± 7.52%, and 90.87% ± 5.68% at 0.1, 1, and 10 μM, respectively (P < .001). Even at high concentrations, corilagin itself displayed no toxicity on the PC12 cell survival without Aβ 25–35 treatment (data not shown).
The protective effects of corilagin on Aβ 25–35 cytotoxicity in PC12 cells were further confirmed by fluorescence-activated cell sorting (FACS) analysis. Similar to the protective effects of corilagin revealed by the MTT assay, pretreatment with corilagin significantly restored cellular viability following exposure to Aβ 25–35, in a dose-dependent manner. The mean cellular viability rate of 70.2% ± 1.78% in Aβ 25–35-treated PC12 cells was increased to 80.3% ± 4.31%, 93.2% ± 1.98%, and 96.0% ± 0.35% by pretreatment with the concentrations of corilagin at 0.1, 1, and 10 μM, respectively (Fig. 1C).
As shown in Figure 1D, after exposure to Aβ 25–35, the intracellular ROS level markedly amplified to 100% ± 2.42% (P < .05) in comparison to the control 69.61% ± 16.88%, which suggests that Aβ 25–35 induces oxidative stress. When the cells were incubated with different concentrations of corilagin (0.1, 1, and 10 μM) in the presence of Aβ 25–35, the intracellular ROS levels significantly decreased to 68.8% ± 8.24%, 56.64% ± 3.67%, and 51.21% ± 5.24%, respectively (P < .01, P < .001, and P < .001, respectively).
Corilagin attenuated Aβ 25–35-induced apoptosis and caspase-3 activation
As presented in Figure 2A, treatment with 50 μM Aβ 25–35 increased DNA fragmentation to 28.33% ± 4.36%, while pretreatment with corilagin apparently decreased fragmented DNA to 18.22% ± 5.49%, 15.01% ± 0.13%, and 8.91% ± 1.39% at 0.1, 1, and 10 μM, respectively (P < .01, P < .001, and P < .001, respectively). Moreover, the protective effect of corilagin at 10 μM was similar to that of resveratrol, the positive control, at 50 μM (5.81% ± 2.09%).
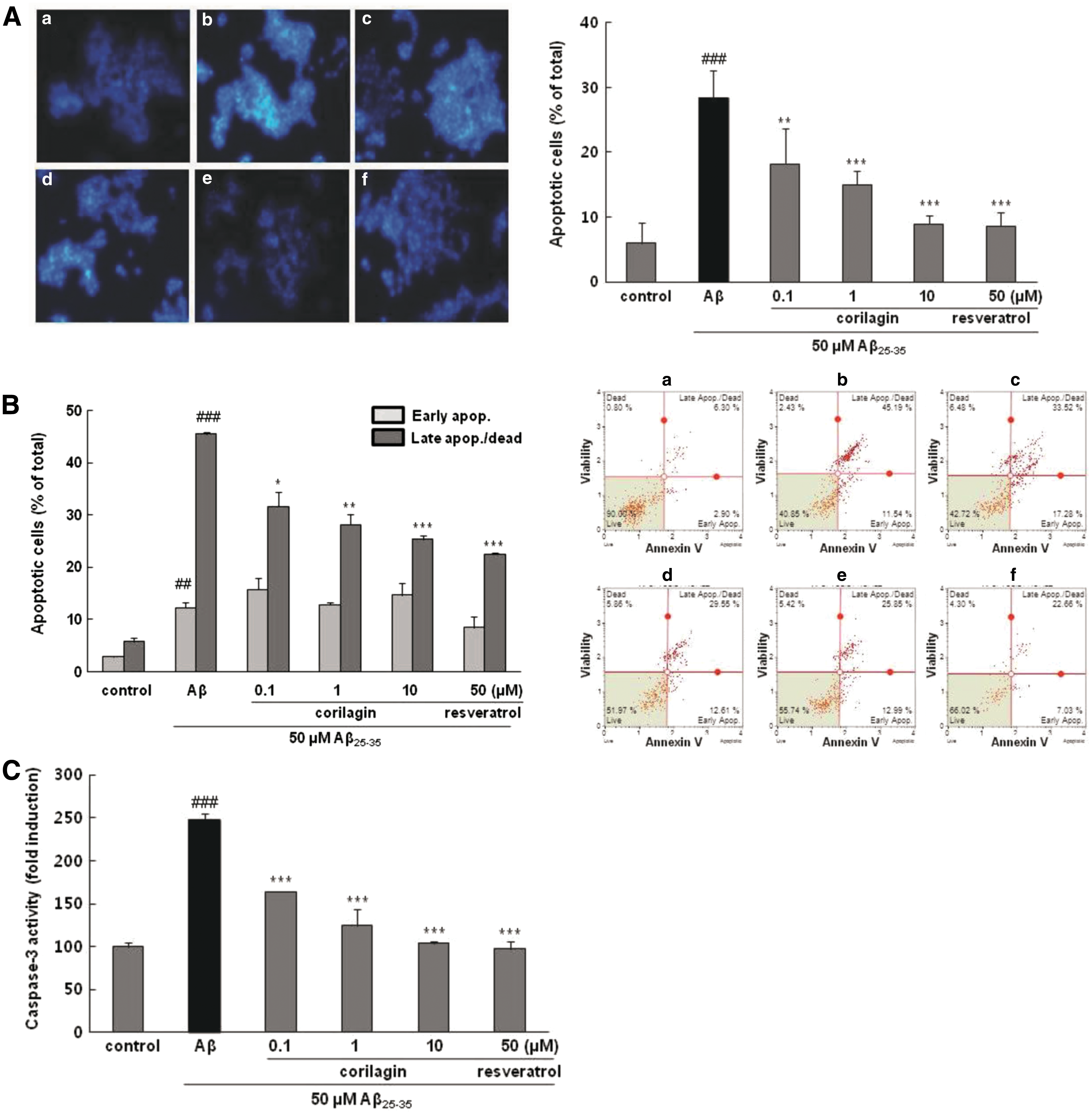
Effects of corilagin on Aβ
25–35 induced cell apoptosis, caspase-3 activation, and disruption of the cell cycle in PC12 cells. The cells were pretreated with corilagin for 1 h and further treated with 50 μM of Aβ
25–35 for 24 h.
For further confirmation of the above results, the protective effect of corilagin on apoptosis of PC12 cells was evaluated using flow cytometry with Annexin V/7-AAD double staining, which distinguishes the early and late stages of apoptosis and provides a quantitative analysis. The percentage of late-stage apoptosis in the control group was 5.75% ± 0.78%, whereas treatment with Aβ 25–35 substantially increased late-stage apoptotic percent to 45.50% ± 0.44% (Fig. 2B, P < .001). Corilagin markedly prevented the late stage of apoptotic rate to 31.60% ± 2.72%, 29.16% ± 1.97%, and 25.39% ± 0.65% at 0.1, 1, and 10 μM, respectively (P < .05, P < .01, and P < .001, respectively). Both manual and FACS results showed that the antiapoptotic property of corilagin might be partially responsible for the decreased cytotoxicity of Aβ 25–35 and improved cell viability in PC12 cells.
Caspase-3, a critical downstream effecter of the cysteine protease family, is the final executor of apoptosis and its activation has been recognized as a main cause of apoptosis. 15 Aβ 25–35 caused an increase in the caspase-3 activity by about 2.3-fold induction compared with that of the control group, while pretreatment of corilagin resulted in a significant attenuation of this effect (P < .001, Fig. 2C).
Corilagin protected Aβ 25–35-induced cell cycle arrest in PC12 cells
The regulatory effect of corilagin on cell cycle by DNA content analysis was further investigated (Fig. 2D). Aβ 25–35 exposure significantly increased the percentage of cells in the G0/G1 phase from 58.7% ± 0.42% to 73.2% ± 0.42% with a concomitant reduction in the percentage of cells in the S phase (decreased from 15.75% ± 2.62% to 11.1% ± 0.99%, P < .001) and G2/M phase (decreased from 25.50% ± 3.11% to 15.70% ± 0.57%, P < .05). Corilagin noticeably suppressed the increase in G0/G1 phase and the decrease in S phase triggered by Aβ 25–35 stimulation, comparable to those of Aβ 25–35-untreated controls. This result suggests that corilagin attenuated Aβ 25–35-provoked cell cycle delaying with the cells accumulating in the G0/G1 phase. The G1 arrest checkpoint may afford a proper time for the repair of DNA damage before the injured cells enter the S phase, which would prevent the translation of damaged DNA into daughter cells. 16
Corilagin inhibited Aβ 25–35-induced TNF-α protein expression
The potential effect of corilagin on the expression of the proinflammatory cytokine TNF-α was determined. The PC12 cells treated with 50 μM Aβ 25–35 alone showed a significant increase in protein concentrations of TNF-α compared with the control (P < .001, Fig. 3). However, corilagin dose dependently inhibited the increase of TNF-α stimulated by Aβ 25–35. Particularly, corilagin at the concentration of 1 and 10 μM almost restored TNF-α expression to the control level.
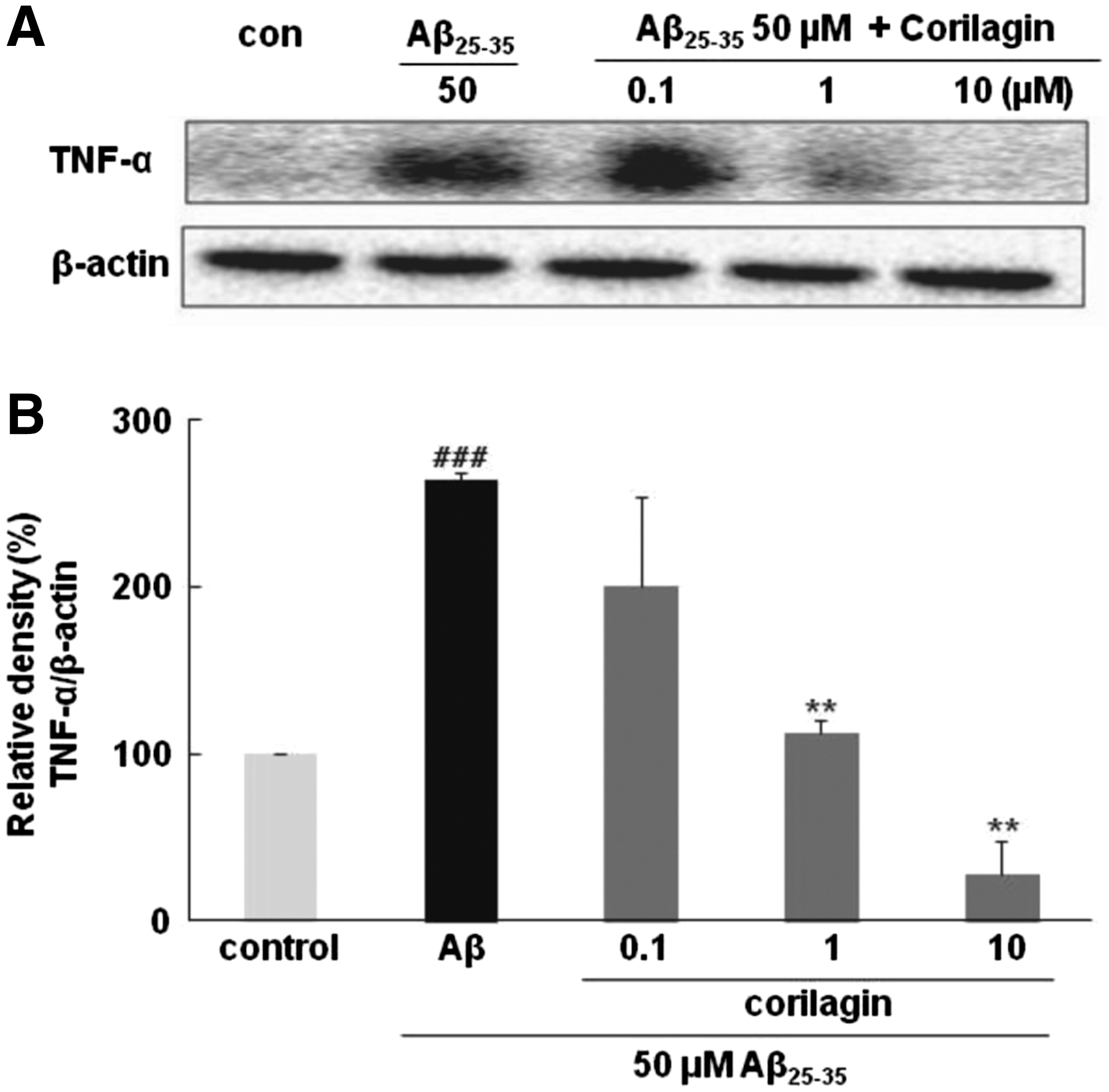
Effects of corilagin pretreatment on Aβ
25–35-induced TNF-α level in PC12 cells.
Corilagin repressed Aβ 25–35-induced productions of NO and PGE2 and expression of iNOS and COX-2
Aβ 25–35-treatment significantly induced NO production (100% ± 11.11%, P < .01) compared to that in the control in Figure 4A. Pretreatment with corilagin at 0.1, 1, and 10 μM distinctly diminished NO levels in PC12 cells exposed to Aβ 25–35, in a dose-dependent manner to 41.5% ± 1.08%, 31.7% ± 1.01%, and 16.2% ± 2.99%, respectively. Consistent with its effect on NO production, corilagin significantly reduced iNOS expression in Aβ 25–35-induced PC12 cells (P < .05, Fig. 4B). Particularly, after preincubation of PC12 cells with 10 μM of corilagin, the level of iNOS decreased down to 79.48%, which implies that the inhibitory property of corilagin on iNOS expression occurred in parallel with the comparable suppression of NO generation.
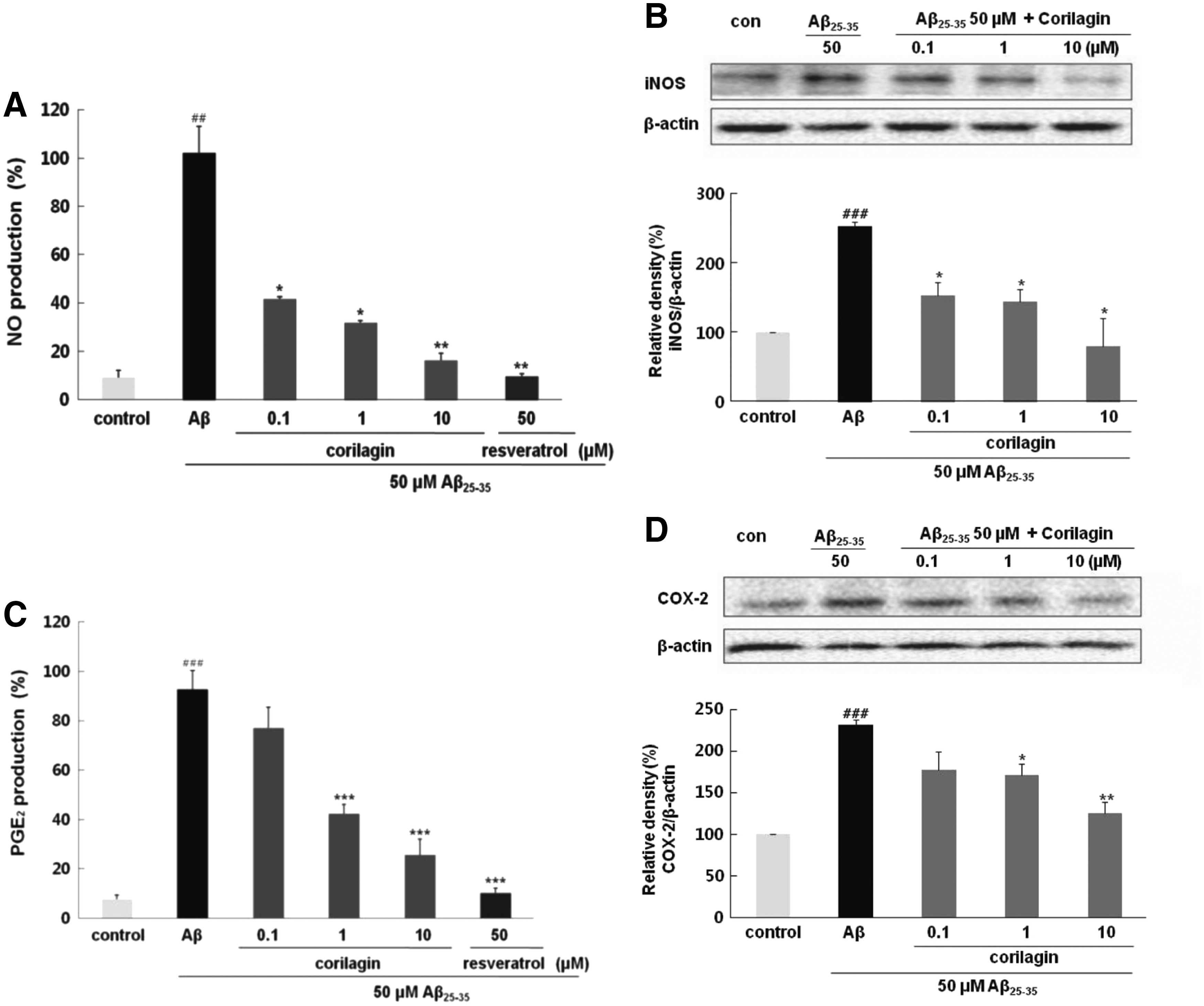
Effects of corilagin pretreatment on Aβ
25–35-induced NO and PGE2 production and iNOS and COX-2 protein expression in PC12 cells.
The effects of corilagin on the production of PGE2 and expression of COX-2 were investigated in PC12 cells treated with Aβ 25–35. As shown in Figure 4C, PGE2 production in response to Aβ 25–35 was significantly inhibited by pretreatment with corilagin in a dose-dependent manner. The maximal inhibition rate of PGE2 production by corilagin was 31.09% ± 6.32% at the concentration of 10 μM (P < .001). These results indicated that the inhibitory effect of corilagin on PGE2 release by Aβ 25–35 was closely regulated by the downregulation of COX-2 in PC12 cells (Fig. 4D).
Corilagin suppressed Aβ 25–35- induced NF-κB and IκB-α expressions
Corilagin is associated with the downregulated expression of iNOS and COX-2 as well as the attenuation of inflammatory cytokines, probably due to the suppression of NF-κB activity. As shown in Figure 5A and B, Aβ 25–35 significantly increased the levels of p65 by 238.58% ± 45.07% in comparison to the control group (P < .01). However, corilagin effectively blocked the phosphorylation of p65 in a dose-dependent manner. In particular, corilagin at 1 and 10 μM displayed almost complete inhibition of p65 phosphorylation to 121.87% ± 31.57% (p < 0.05) and 94.36% ± 17.44% (P < .01), respectively. Similar to the extent of p65 phosphorylation, Aβ 25–35 markedly increased the expression of phospho-IκB-α, which was significantly suppressed by corilagin at 1 and 10 μM (P < .05 and P < .01, Fig. 5C). These findings clearly revealed that attenuation on NF-κB activation by corilagin probably led to repression of TNF-α, NO, PGE2, iNOS, and COX-2.
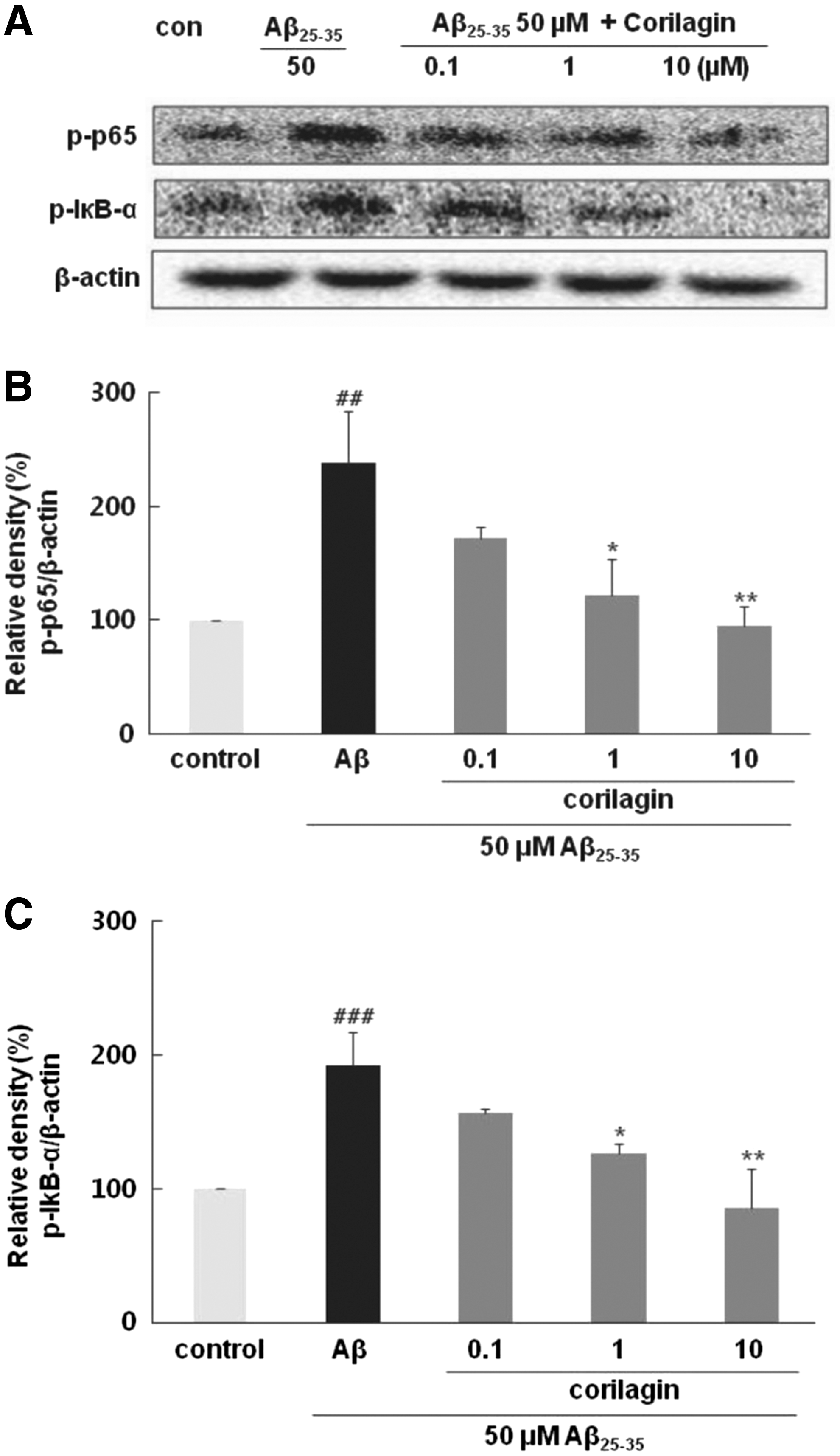
Effects of corilagin pretreatment on Aβ
25–35-induced NF-κB and IκB-α activation in PC12 cells.
Corilagin induced downregulation of Aβ 25–35-induced MAPK signaling
Treatment with Aβ 25–35 prominently increased the degree of phosphorylation of p38, ERK ½, and JNK to 194.26% ± 2.04%, 551.80% ± 39.23%, and 264.23% ± 40.10% in comparison to the control values (P < .001, P < .001, and P < .01, respectively). Interestingly, among the major upstream kinases, corilagin exhibited more potent suppression against ERK1/2 than p38 or JNK (Fig. 6). Corilagin led to significant downregulation phosphorylation of ERK1/2 to 105.74% ± 28.16%, 75.28% ± 19.35%, and 54.70% ± 8.55% at concentrations of 0.1, 1, and 10 μM, respectively (P < .001). In contrast to the insignificant decrease in Aβ 25–35-induced p38 and JNK phosphorylation levels at 0.1 and 1 μM of corilagin, 10 μM of corilagin showed a relatively significant decrease in the phosphorylation of p38 and JNK to 82.49% ± 32.79% and 120.53% ± 26.57%, respectively (P < .05 and P < .01). These data suggested that corilagin possibly regulates the inhibition of Aβ 25–35-stimulated PC12 cells mostly through the suppression of ERK 1/2 expression.
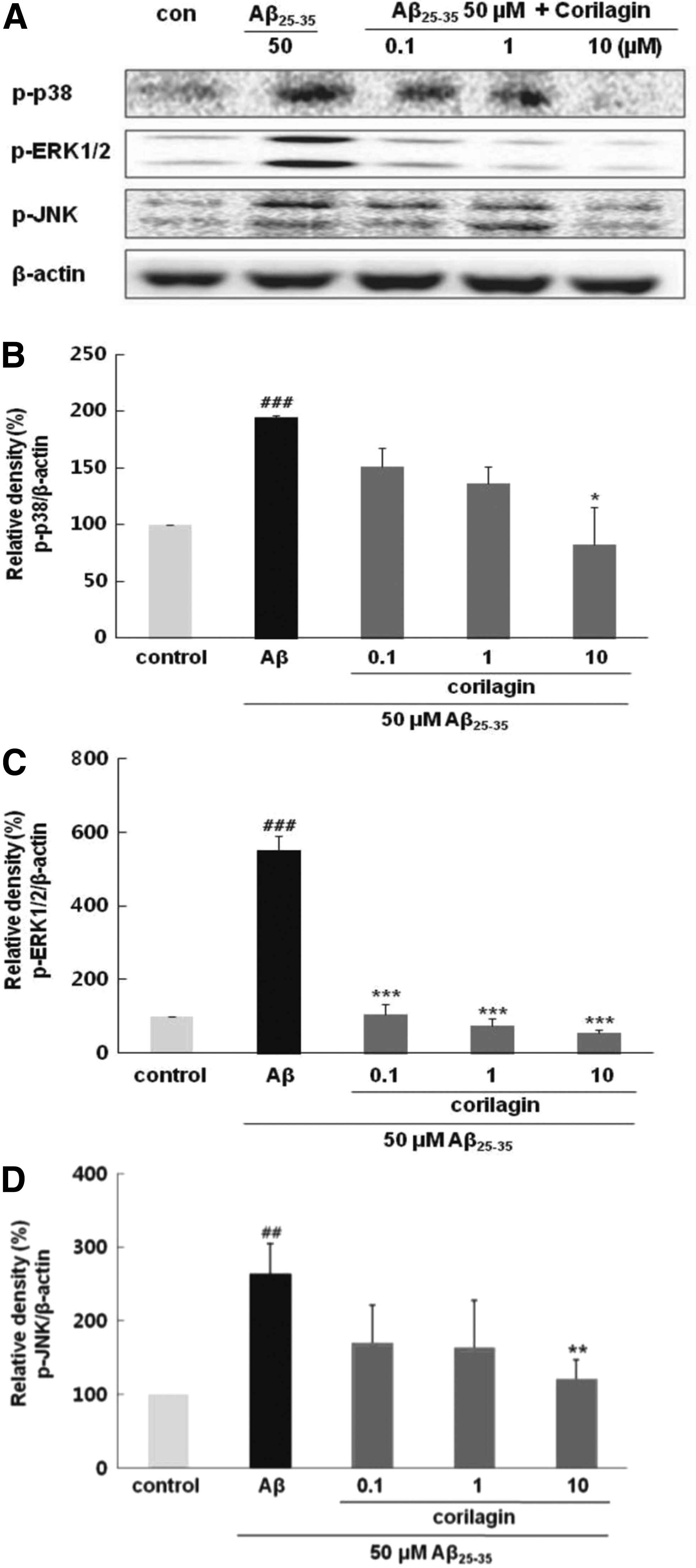
Effects of corilagin pretreatment on Aβ
25–35-induced MAPK phosphorylation in PC12 cells.
Discussion
The human brain is a vital organ, more prone to oxidative stress than any other organ, owing to its high metabolic rate and polyunsaturated fatty acid content. Even though the specific mechanisms of Aβ-induced neurotoxicity are not fully understood, several possible mechanisms by which Aβ triggers neurotoxicity have been proposed and one of them being oxidative stress accelerated by Aβ. 17 Increased oxidative damage has been reported in the early stages of cognitive impairment in the brains of individuals with AD, and destruction of antioxidant defense system was demonstrated in a number of models. 18 –20 The brains of individuals with AD had increased levels of lipid peroxidation products, including 4-hydroxy-nonenal and/or 2-propenal, and enhanced lipid peroxidation has also been detected in the cerebrospinal fluid and plasma of individuals with AD. 21 –23 In this study, corilagin suppressed Aβ 25–35-dependent damage, to some extent by reducing oxidative stress, noticeably demonstrated by the decreased levels of ROS and NO, resulting in restoration of cellular viability. Several groups have demonstrated that corilagin attenuates tert-butyl hydroperoxide-stimulated oxidative injury in microglial cells and GalN/ lipopolysaccharide (LPS)-induced liver injury through suppression of oxidative stress and apoptosis in rat. 24,25
Another neurotoxic mechanism exhibited by Aβ 25–35 is neuroinflammation. Results of this study demonstrated that corilagin could inhibit production of TNF-α, PGE2, and NO by downregulating the expression of iNOS and COX-2. In accordance with our results, corilagin was reported to be an inhibitor of TNF-α in BALB/3T3 cells and decreased carrageenan-induced hyperalgesia through inhibition of PGE2 production. 26,27 Furthermore, regulation of NF-κB signaling pathway by corilagin in Aβ 25–35-damaged PC12 cells implies involvement and restoration of this pathway by corilagin. Similar inhibitory results of corilagin against NF-κB activation have been observed in several different cell types, including microglia, RAW264.7, intestinal epithelial cells, epithelial cells in patients with cystic fibrosis, and IB3-1 cells exposed to various inflammatory stimuli such as radiation, LPS, herpes simplex virus (HSV)-1, dextran sulfate sodium, and TNF-α. 10,28 –31
Based on our results that corilagin suppressed Aβ 25–35-triggered NF-κB activation, further effects of corilagin on upstream signaling pathways were examined. The signal transduction pathways involved in Aβ-dependent neurotoxicity have become a major focus in current AD research. MAPKs, including at least three families of MAPK (ERK 1/2, JNK, and p38), can be stimulated by Aβ, which undergo phosphorylation and activation of transcription factors during inflammatory responses. 32 In this study, corilagin was verified as a potent suppressor of MAPKs, especially of ERK1/2, which then regulated the expression of iNOS and COX-2 through the modulation of NF-κB in Aβ 25–35-treated PC12 cells.
In recent years, over 50 different kinds of plants or herbs, either in the form of pure compounds or extracts, have been identified to be potentially useful in prevention or treatment of AD. 33 Searching for the health benefits of these natural products poses substantial challenges to modern medicine, especially with their superior safety profiles. 34 Corilagin exhibited efficacy for alleviating brain inflammatory changes, and neuronal injury after HSV-1 infection in Balb/male mice brain. 28 In addition, Park et al. revealed that, oral administration of Terminalia chebula seeds containing corilagin protected the hippocampal C1 region of gerbils from ischemic damage. 35 Furthermore, a recent study by Tong et al. found that administration of corilagin improved spatial learning and memory ability in radiation-induced rat brain injury. 36
Corilagin is a simple ellagitannin with one glucose unit esterified with hexahydroxydiphenic acid (HHDP) and gallic acid moieties. Previous studies on the analysis of active ellagitannin structures demonstrated that an HHDP moiety or its oxidatively modified entities might be the key structural feature resulting in activities like anticryptococcal and antioxidative effects (Latté and Kolodziej, 37 Yokozawa et al., 38 and Rangkadilok et al. 39 ). This key structural feature may also be responsible for anti-inflammatory properties of corilagin in this study. However, little is proven about the absorption, distribution, metabolism, and bioavailability of polymeric tannins and the findings are still controversial. 40 Although a recent study by Marín et al. suggested that ellagitannins undergo lactonization yielding smaller compounds by the colonic microorganisms, there exists no adequate research to verify or contradict that corilagin might be absorbed and metabolized to smaller compounds by intestinal microflora and that it might act as anti-AD agents in the brain. 41 An encouraging finding was that rat plasma corilagin was measured to be maximum 3 h after administration without degradation, which needs to be verified in future studies for not only corilagin itself but also for other tannin compounds. In addition, oral administration of tannin augmented in vivo intrahippocampal antioxidant ability of rats. 42
Corilagin cannot currently be regarded as an anti-AD agent because of its relatively high hydrophilicity and large molecular weight. However, a promising discovery has revealed that large hydrophilic catechins penetrate the BBB and exhibit strong neuroprotective properties. 43 Although further and more detailed explanations on the possibility of its bioavailability and permeability through the BBB are required in the near future, it is still meaningful that corilagin exerted marked neuroprotective effects against Aβ 25–35-triggered neurotoxicity. Further in vivo study of this activity is required to elaborate the mechanisms and understand its promise, especially to alter signal transduction.
Footnotes
Acknowledgment
This research was supported by Dong-A University.
Author Disclosure Statement
No competing financial interests exist.