
Abstract
Sargassum serratifolium was found to contain high concentrations of meroterpenoids, having strong antioxidant, anti-inflammatory, and neuroprotective activities. This study aims to investigate the anti-inflammatory mechanisms of an ethanolic extract of S. serratifolium (ESS) using lipopolysaccharide (LPS)-stimulated BV2 microglial cells and to identify the anti-inflammatory components in ESS. The level of proinflammatory cytokines was measured by enzyme-linked immunosorbent assay. The expression of inflammation-related proteins and mRNA was evaluated by Western blot and reverse transcription-polymerase chain reaction analysis, respectively. Anti-inflammatory activities of isolated components from ESS were analyzed in LPS-stimulated BV2 cells. ESS inhibited LPS-induced nitric oxide (NO) and prostaglandin E2 and the expression of inducible NO synthase and cyclooxygenase-2. ESS also decreased the release of proinflammatory cytokines in a dose-dependent manner. LPS-induced nuclear factor-kappa B (κB) transcriptional activity and translocation into the nucleus were remarkably suppressed by ESS through the prevention of inhibitor κB-α degradation. The main anti-inflammatory components in ESS were identified as sargahydroquinoic acid, sargachromenol, and sargaquinoic acid based on the inhibition of NO production using LPS-stimulated BV2 cells. Furthermore, treatment with ESS significantly reduced levels of tumor necrosis factor-α and interleukin-1β stimulated with LPS in mouse hippocampus. Our results indicate that ESS can be used as a functional food or therapeutic agent for the treatment of neuroinflammatory diseases.
Introduction
M
The production of proinflammatory cytokines and proteins is mainly regulated by nuclear factor-kappa B (NF-κB). 7 NF-κB plays a critical role in the regulation of inflammation. NF-κB is bound to inhibitory κB (IκB) in the cytoplasm as an inactive form. Lipopolysaccharide (LPS)-induced phosphorylation of IκB kinase (IKK) leads to phosphorylation and degradation of IκB, and thus resulting in free NF-κB translocating into the nucleus 8 where it induces transcriptional regulation of inflammatory genes. 9,10 NF-κB is also activated by protein kinases such as mitogen-activated protein kinases (MAPKs). MAPKs include extracellular signal-regulated kinases (ERKs), p38 MAPK, and c-Jun NH2-terminal kinases (JNKs). 11 The phosphorylation of MAPKs induces NF-κB activation. Thus, the blockade of MAPK phosphorylation can suppress the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) via inhibition of the NF-κB pathway.
Sargassum serratifolium is a perennial brown alga of the Sargassaceae family and is widely distributed on the Korean and Japanese coasts. Recently, we found that the ethanolic extract of S. serratifolium (ESS) contains a high level of meroterpenoids and isolated sargaquinoic acid, which showed high anti-inflammatory activities in RAW 264.7 and human umbilical vein endothelial cells. 12,13 Thus, we investigated the anti-inflammatory mechanisms of ESS using LPS-stimulated BV2 microglial cells and identified the anti-inflammatory compounds in ESS.
Materials and Methods
Materials
LPS (Escherichia coli O55:B5), dimethyl sulfoxide (DMSO), 4′,6-diamidino-2-phenylindole (DAPI), and protein kinase inhibitors (SB203580, SP600125) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM), penicillin–streptomycin mixture, and fetal bovine serum (FBS) were obtained from Gibco-BRL Life Technologies (Grand Island, NJ, USA). CellTiter96 Aqueous One Solution Cell Proliferation Assay Kit, Dual Luciferase Assay Kit, murine NF-κB promoter/luciferase DNA, pRL-TK DNA, and Moloney murine leukemia virus reverse transcriptase were obtained from Promega (Madison, WI, USA). Primary and secondary antibodies were purchased from Cell Signaling Biotechnology (Danvers, MA, USA). LipofectAMINE PLUS Reagent and TRIzol reagent were purchased from Invitrogen (Carlsbad, CA, USA). Enzyme-linked immunosorbent assay (ELISA) kits were obtained from R&D Systems (Minneapolis, MN, USA).
Plant materials
S. serratifolium was collected along the coast of Busan, Republic of Korea, in July 2014. Specimens were identified by an algal taxonomist (C.G. Choi) at the Department of Ecological Engineering, Pukyong National University, Republic of Korea. The samples were dried for 2 days and ground with hammer grinder. Dried power (1.0 kg) of S. serratifolium was extracted two times with 95% (v/v) ethanol for 3 h at 70°C. The combined ethanolic extracts were concentrated using a rotary vacuum evaporator (Eyela, Tokyo, Japan) at 40°C and lyophilized to remove water (105 g). Aliquot of ESS was dissolved in DMSO and further diluted with culture media before use. The final concentration of DMSO in cell culture medium was less than 0.1%.
Isolation and identification of anti-inflammatory compounds from ESS
The HPLC system consisted of a photodiode array detector (Shimadzu SPD-M20A), a pump (Shimadzu LC-6AD), an online degasser (Shimadzu DUG-20A3), an autosampler (SIL-20A), a system controller (CBM-20A), a fraction collector (Shimadzu FRC-10A), and a Shimadzu LCsolution (ver.1.22sp). Aliquots of ESS were dissolved in methanol and separated by HPLC system with Luna RP-18 [Luna C18(2), 5 μm, 250 × 10 mm; Phenomenex, Torrence, CA, USA]. The separation of ESS was conducted using 0.1% formic acid in methanol (solvent A) and 0.1% formic acid in water (solvent B) as a mobile phase. The elution profile consisted of a linear gradient from A/B (78/22) to A/B (95/5) for 88 min, A/B (95/5) to A/B (100/0) for 2 min, and re-equilibrated with A/B (78/22) for 20 min after washing for 25 min with A/B (100/0). The flow rate was 3.5 mL/min and peaks were detected at 270 nm. Each fraction was collected and chemical structures were determined by 1H-NMR and 13C-NMR spectra using a JNM ECP-400 (Jeol, Tokyo, Japan). HPLC fingerprint was obtained by the same HPLC system with Luna RP-18 [Luna C18(2), 3 μm, 150 × 3.0 mm]. Analysis was carried out under the same chromatographic conditions at a flow rate of 0.34 mL/min and the injection volume was 3 μL.
Cell cultures and sample treatment
The murine BV2 microglial cells (ATCC, Rockville, MD, USA) were maintained in DMEM supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin sulfate (100 μg/mL) in a humidified atmosphere of 5% CO2. Cells were stimulated with LPS (1 μg/mL) in the presence or absence of ESS for the indicated time.
Cell viability and proliferation
Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay using the CellTiter96 Aqueous One Solution Cell Proliferation assay kit according to the manufacturer's instruction. Cells were inoculated at a density of 1 × 105 cells into 96-well plates and cultured at 37°C for 24 h. The culture medium was removed and replaced by 95 μL of fresh culture medium and 5 μL of MTS solution. After 1 h, the absorbance was measured at 490 nm using a microplate reader.
Measurement of NO, PGE2, and cytokines
Cells (5 × 104 cells/well) were pretreated with ESS (0–2.0 μg/mL) for 2 h before LPS stimulation for 24 h. After stimulation with LPS, cultured media were collected and stored at −75°C until tested. For the measurement of NO, 100 μL of culture media were mixed with the same volume of Griess reagent and incubated at room temperature for 10 min. Absorbance of the mixture was measured with a microplate reader at 540 nm. Levels of PGE2, TNF-α, and IL-6 in cultured media were quantitatively determined by ELISA kit according to the manufacturer's instructions.
COX-2 inhibitory activity assay
The COX-2 activity was analyzed using a COX Inhibitor Screening Assay Kit purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Recombinant COX-2 protein was preincubated with ESS and COX-1 inhibitor (SC-560) for 10 min at 37°C. The reaction was initiated by the addition of 100 μM arachidonic acid and allowed to proceed for 2 min. The reaction was terminated by adding HCl solution containing SnCl2. The COX-2 activity was directly measured as PGF2α produced by SnCl2 reduction of COX-derived PGH2. The prostanoid product was quantified via ELISA. As a negative control, COX-2 was inactivated by boiling for 5 min. Celecoxib was used as a positive COX-2 control.
Reverse transcription-polymerase chain reaction
BV2 cells inoculated in a six-well cell culture plate at a density of 3.0 × 105 cells/well were pretreated without or with ESS for 1 h and then treated with LPS for 6 h. Total RNA was isolated with the TRIzol reagent. Five micrograms of total RNA was used for reverse transcription using oligo-dT and M-MLV reverse transcriptase. Polymerase chain reaction (PCR) was carried out using the resulting cDNA as a template under the following conditions: denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec, and extension at 72°C for 30 sec. The PCR products were visualized by agarose gel electrophoresis. Verification of PCR product was established by their predicted sizes under an ultraviolet light illuminator. GAPDH was used as an internal standard to evaluate relative expression of mRNAs. The primer sequences are shown in Table 1. Densitometric analysis of the data obtained from at least three independent experiments was performed using a cooled CCD camera system EZ Capture II and CS analyzer ver. 3.00 software.
NF-κB promoter/luciferase assay
Two micrograms of pNF-κB promoter/luciferase DNA along with 40 ng of control pRL-TK DNA was transfected onto 2.0 × 105 BV2 cells per well in a six-well plate using LipofectAMINE PLUS reagents for 40 h. Cells were treated with ESS for 2 h and stimulated with LPS (1 μg/mL) for 6 h. Luciferase activities of the cells were measured using a dual-luciferase assay system according to the manufacturer's instruction. The luciferase activity was normalized with control pRL-TK.
Preparation of cytosolic and nucleus extracts
BV2 cells (7 × 105) plated in a 60 mm cell culture plates were pretreated with or without ESS for 2 h and further stimulated with LPS for 30 min. Separation of cytosolic acid nucleus extracts was performed as previous described. 14
Western blot
BV2 cells (5 × 104 cells/well) were pretreated with ESS (0–2.0 μg/mL) for 2 h before LPS stimulation at indicated times in the figure legends. The specific procedure for Western blotting is described in our previous article. 14
Immunocytochemical analysis
Cells were cultured on glass coverslips (SPL Lifesciences Co., Gyeonggi-do, Korea) in six-well plates for 24 h. After pretreatment with indicated concentrations of ESS for 2 h, cells were stimulated with LPS (1 μg/mL) for 30 min. The specific procedure for immunofluorescence analysis of NF-κB is described in our previous article. 14
Animal study
Male ICR mice (6 weeks) were obtained from Orient Bio Co. (Gyeonggi-do, Korea). Mice were adapted to the animal laboratory unit for 1 week before experiments. Mice were randomly divided into four groups of five mice each. Group I (control group) was orally administrated with normal saline for 4 days and saline was intraperitoneally injected at 2 h after last administration. Group II (negative group) was administrated with saline for 4 days and LPS (2 mg/mL) was intraperitoneally injected at 2 h after last administration. Group III was administrated with ESS (10 mg/kg) for 4 days and LPS (2 mg/mL) was intraperitoneally injected at 2 h after last administration. Group IV was administrated with ESS (20 mg/kg) for 4 days and LPS (2 mg/mL) was intraperitoneally injected at 2 h after last administration. After 2 h post-LPS or saline stimulation, hippocampi were collected from mice sacrificed by cervical dislocation. Obtained hippocampi were homogenized in RIPA buffer and lysate was obtained after centrifugation (12,000 g, 5 min). Concentrations of TNF-α and IL-1β in lysates were determined with the ELISA kit.
Statistical analysis
Data are expressed as means ± standard deviations of at least three independent experiments unless otherwise indicated. Data were analyzed using one-way analysis of variance (ANOVA), followed by each pair of Student's t-test for multiple comparisons. Differences with a value of P < .05 were considered statistically significant.
Results
ESS inhibits LPS-induced NO and PGE2 production
As shown in Figure 1A, ESS pretreatment for 2 h decreased NO production in LPS-stimulated BV2 (Fig. 1A) in a dose-dependent manner. Moreover, increased levels of PGE2 by LPS stimulation were markedly lower at low concentrations of ESS in BV2 (Fig. 1B). ESS has no cytotoxicity up to 2.0 μg/mL concentration (Fig. 1C).
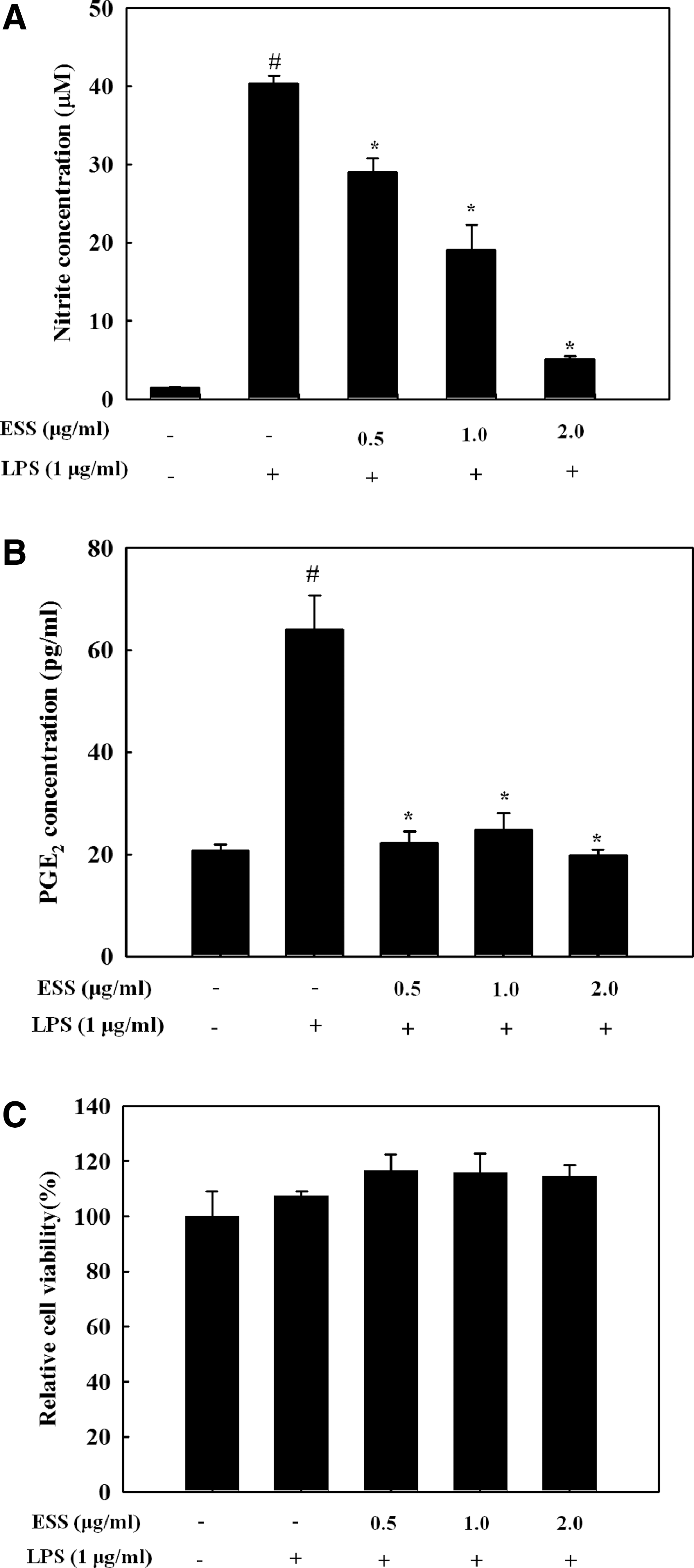
Effect of ESS on the production of NO and PGE2 in LPS-stimulated BV2 microglial cells. BV2 microglial cells pretreated with indicated concentrations of ESS for 2 h were stimulated with LPS (1 μg/mL) for 24 h. The culture media were used to determine NO
ESS suppresses LPS-induced iNOS and COX-2
Since iNOS and COX-2 are responsible for the production of NO and PGE2, respectively, we assessed the suppressive effect of ESS on the expression of both protein and mRNA. As shown in Figure 2A, iNOS protein and mRNA remarkably reduced by ESS in a dose-dependent manner. However, the COX-2 protein and mRNA reduction by ESS was less than iNOS in LPS-stimulated BV2 cells. The decrease in COX-2 protein and mRNA was quite low compared to that of PGE2, which suggests that ESS has COX-2 inhibitory activity. Thus, we determined the COX-2 inhibitory activity of ESS. As shown in Figure 2B, ESS inhibited COX-2 activity in a dose-dependent manner. Thus, the decreased production of PGE2 by ESS was caused by direct inhibition of COX-2 activity, not COX-2 expression.
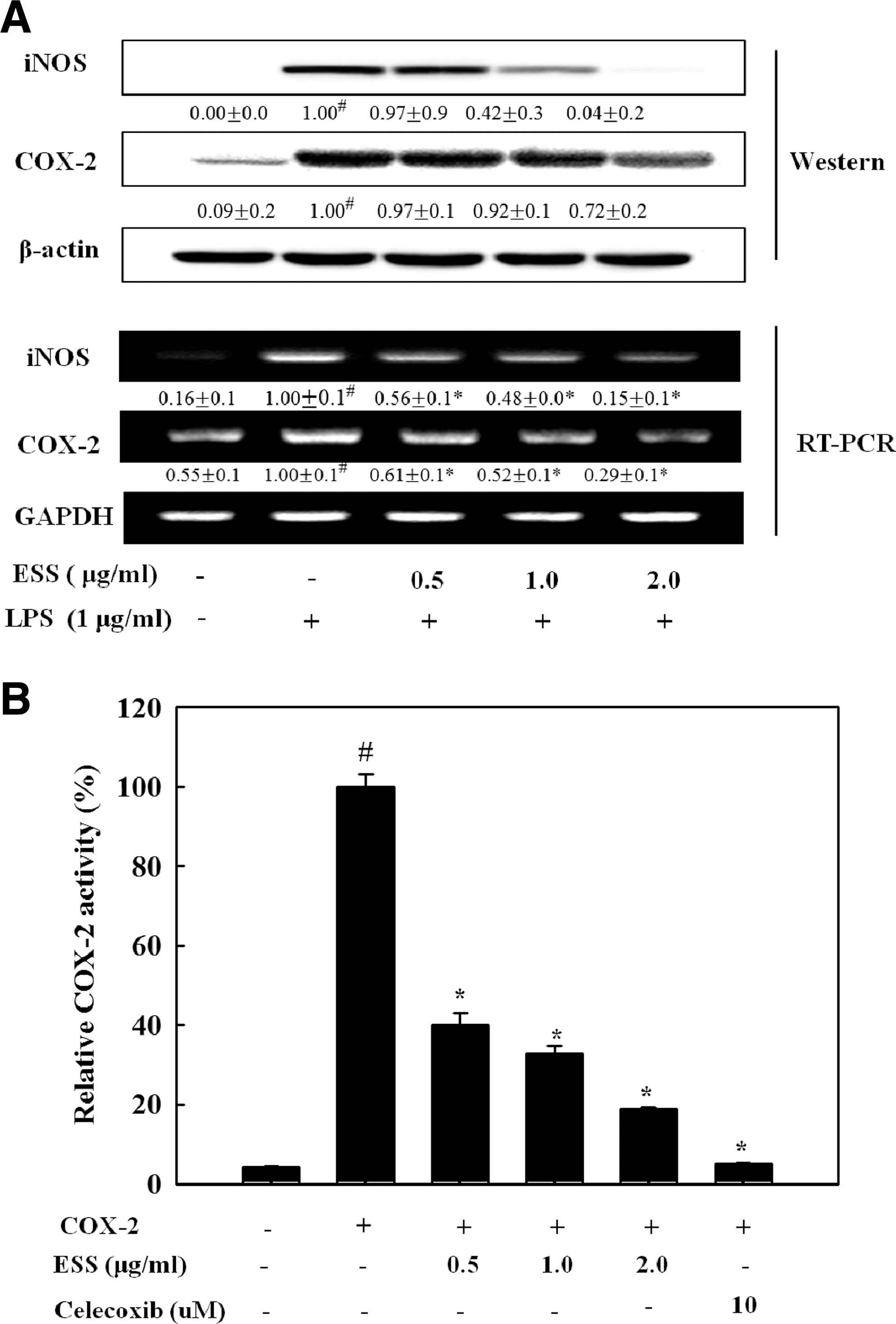
Effect of ESS on iNOS and COX-2 protein and mRNA expressions in LPS-stimulated BV2 microglial cells. Cells were pretreated with the indicated concentrations of ESS for 2 h and stimulated with LPS (1 μg/mL) for 16 h for Western blot or stimulated with LPS (1 μg/mL) for 6 h for RT-PCR
ESS suppresses LPS-induced IL-6 and TNF-α production
To determine the effect of ESS on the levels of TNF-α and IL-6, BV2 cells were pretreated with ESS for 2 h and further stimulated with LPS for 24 h. The levels of TNF-α (Fig. 3A) and IL-6 (Fig. 3B) in the medium were significantly lower with ESS pretreatment (P < .05). Moreover, mRNA levels of TNF-α and IL-6 were inhibited by ESS in LPS-stimulated BV2 cells (Fig. 3C).
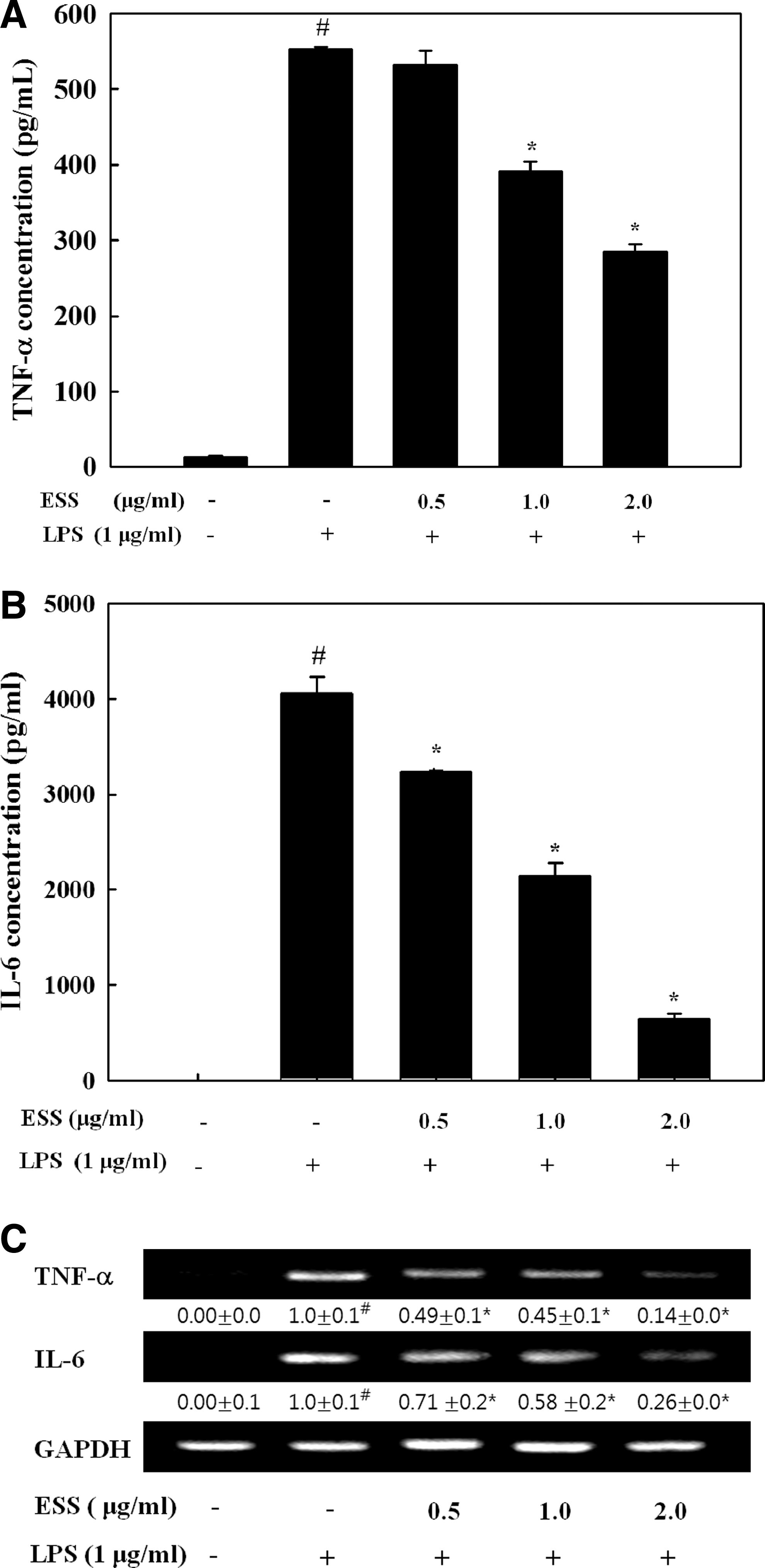
Effect of ESS on secretion of proinflammatory cytokines in LPS-stimulated BV2 microglial cells. Cells were pretreated with indicated concentrations of ESS for 2 h and stimulated with LPS (1 μg/mL) for 24 h. Culture media and cells were separated. TNF-α
ESS inhibits LPS-induced NF-κB activation
To assure whether ESS inhibits NF-κB activation in LPS-stimulated BV2 cells, the effect of ESS on NF-κB translocation was assessed by confocal microscopy. Immunofluorescence analysis (Fig. 4A) indicated that NF-κB in the cytoplasm was largely translocated into the nucleus by LPS stimulation. However, ESS pretreatment (2.0 μg/mL) blocked its translocation in LPS-stimulated BV2 cells. To confirm the molecular mechanism of ESS on the blockade of NF-κB translocation, we analyzed the level of proteins associated with NF-κB translocation by Western blot. LPS induced the phosphorylation of IKKβ, which is responsible for IκBα phosphorylation; however, ESS dose dependently inhibited IKKβ phosphorylation. The inhibition of IKKβ led to the suppression of IκBα phosphorylation and restored the NF-κB level in cytosol (Fig. 4B). In addition, the enhanced NF-κB level in nuclear fraction by LPS was suppressed by ESS in a dose-dependent manner. Moreover, NF-κB promoter activity assay using luciferase construct indicated remarkable inhibition of NF-κB promoter activity by ESS (Fig. 4C). These results suggest that ESS-mediated suppression of inflammatory responses was mainly regulated by the inhibition of NF-κB activation in LPS-stimulated BV2 cells.

Effect of ESS on IκB phosphorylation and NF-κB activation in LPS-stimulated BV2 microglial cells. Cells pretreated with indicated concentrations of ESS for 2 h were stimulated with LPS (1 μg/mL) for 30 min. Cells were stained by anti-NF-κB/p65 antibody and DAPI, and then, immunofluorescence was observed by confocal microscopy ( × 40)
ESS suppresses LPS-induced JNKs and p38 MAPK phosphorylation
Since NF-κB activation is alternatively modulated by intracellular signaling proteins, MAPKs, in LPS-exposed BV2 cells, we further examined whether ESS regulates the phosphorylation of these signaling proteins activated by LPS in BV2. As shown in Figure 5A, ESS suppressed the phosphorylation of JNK1/2 and p38 MAPK in LPS-stimulated BV2 cells, indicating the additional characteristics of ESS to regulate activation of NF-κB in response to LPS signal. To confirm the association with these signaling proteins with the ESS anti-inflammatory effect, we analyzed the production of NO in the presence of ESS, JNK inhibitor (SP600125), and p38 MAPK inhibitor (SB203580). As shown in Figure 5B, NO production from the LPS-stimulated BV2 cells was markedly decreased by ESS as well as by JNK and p38 MAPK inhibitors. These results suggest that there are additional properties of ESS to regulate the NF-κB pathway via blocking the phosphorylation of JNKs and p38 MAPK proteins in response to LPS.
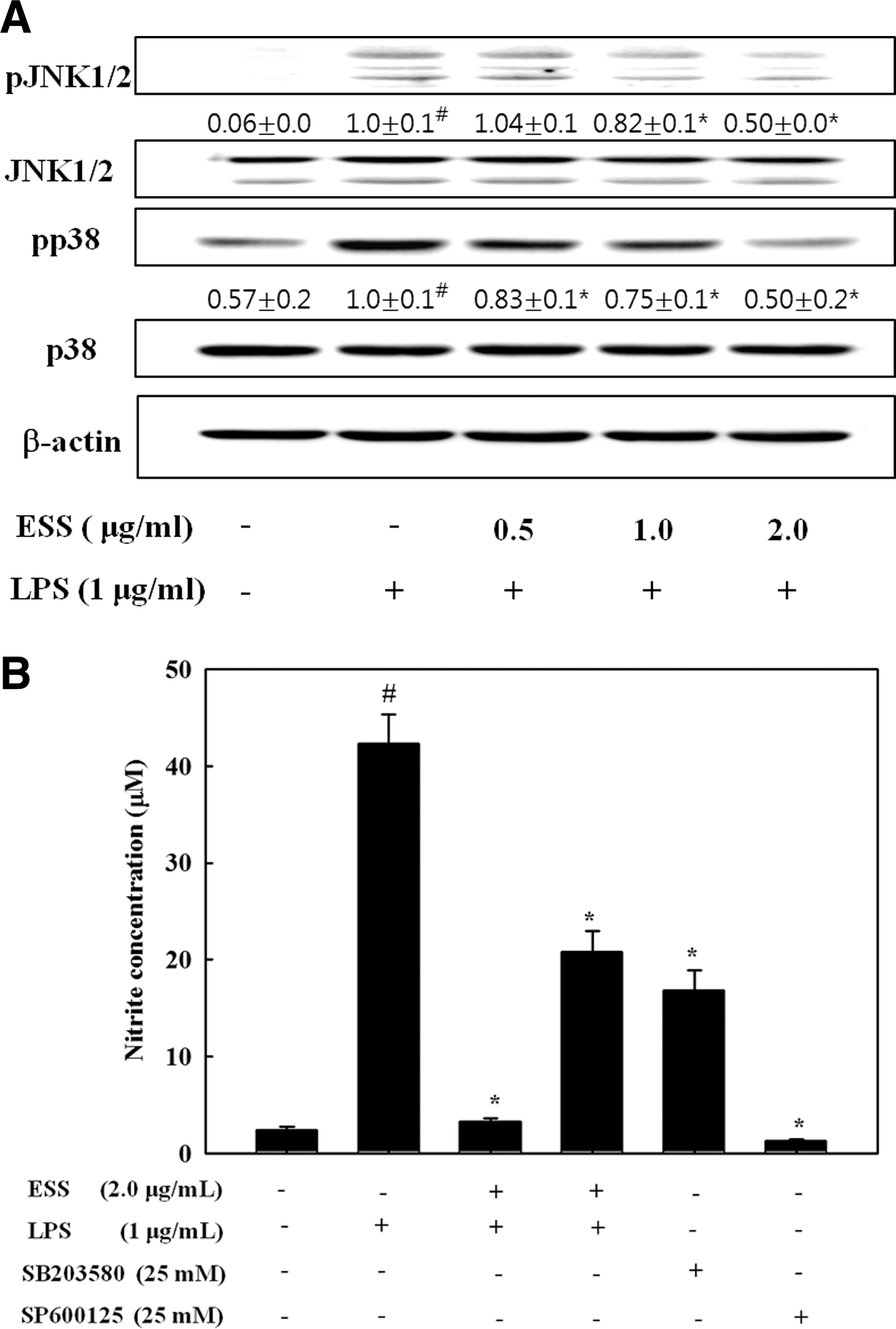
Effect of ESS on the phosphorylation of JNK1/2 and p38 MAPK in LPS-stimulated BV2 microglial cells. BV2 cells pretreated with indicated concentrations of ESS for 2 h were stimulated with LPS (1 μg/mL) for 30 min. The cell lysates were measured by Western blotting for total and phosphorylated proteins of JNK1/2 and p38 MAPK, respectively
ESS suppresses inflammatory cytokines in hippocampus
To assess effects of ESS in vivo on inflammatory responses, mice administrated with ESS for 4 days were stimulated with LPS (i.p) for 2 h and cytokine levels were determined in mouse hippocampi. As shown in Figure 6, the increased levels of TNF-α and IL-1β by LPS were significantly reduced by 20 mg/kg administration of ESS (P < .05), whereas 10 mg/kg ESS treatment did not show a significant effect on TNF-α and IL-1β release.
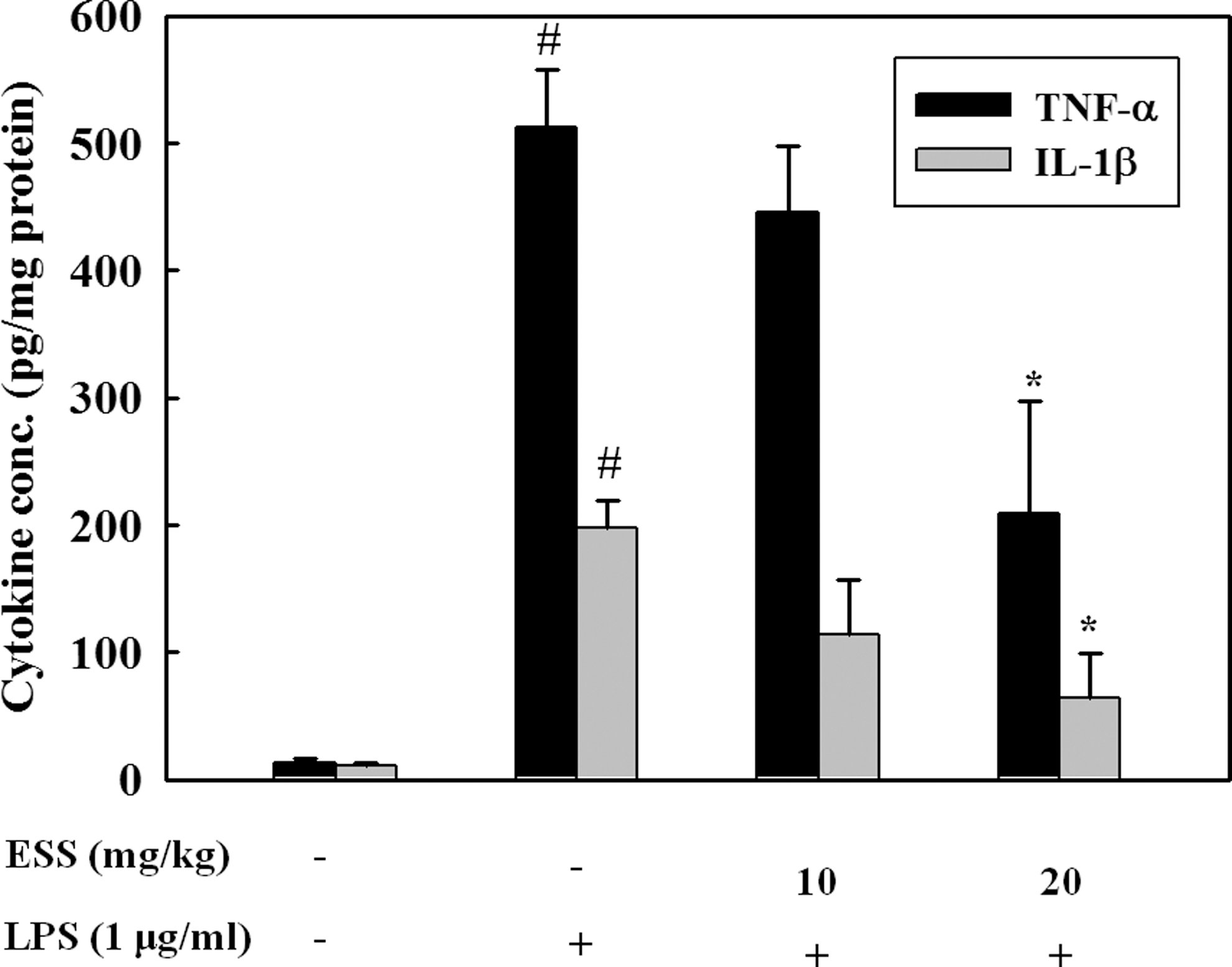
Effect of ESS administration on the inhibition of proinflammatory cytokines in LPS-challenged mice. TNF-α and IL-1β levels in the hippocampus were determined after 2 h of LPS stimulation by ELISA. # P < .05 indicates significant differences from the control group. *P < .05 indicates significant differences from the LPS-only group.
Isolation and identification of anti-inflammatory compounds from ESS
The HPLC fingerprint of ESS gave separated four peaks at different retention times (Fig. 7). The peaks were collected by the repeated chromatographies, and the chemical structure of the isolated compound was identified by 1D (1H and 13C NMR) and 2D (heteronuclear multiple-quantum correlation and heteronuclear multiple-bond correlation.) spectroscopic analyses. In this study, we isolated three compounds and sargahydroquinoic acid, sargachromenol, and sargaquinoic acid were identified by their NMR spectroscopies. Anti-inflammatory activities of sargahydroquinoic acid, sargachromenol, and sargaquinoic acid were determined by inhibition of NO production using LPS-stimulated BV2 cells. The EC50 of sargahydroquinoic acid, sargachromenol, and sargaquinoic acid on the inhibition of NO production was estimated to be 0.21 ± 0.02, 1.26 ± 0.11, and 0.47 ± 0.05 μM, respectively (Table 2).
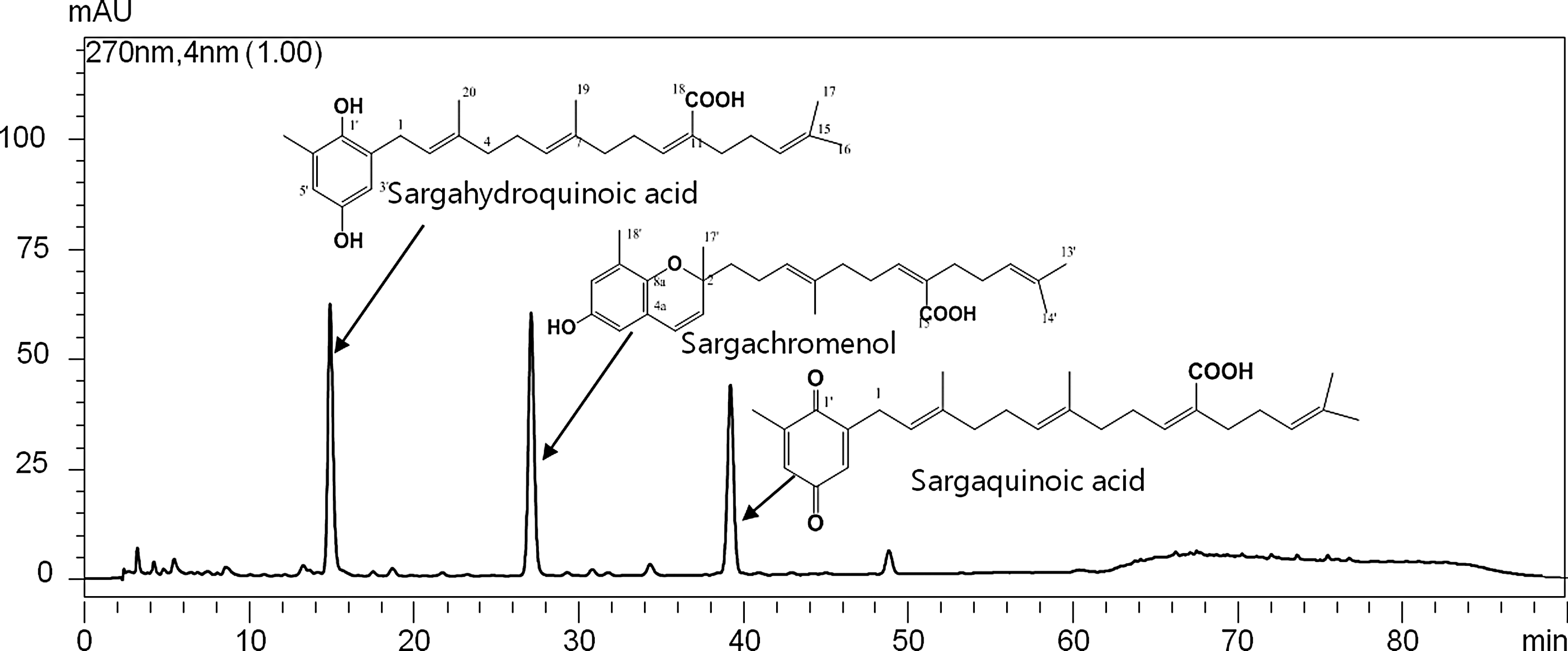
Representative HPLC chromatogram of ESS. Chemical structures of the separated compounds were elucidated by NMR spectroscopy.
EC50 was measured by inhibitory activity of NO production in LPS-induced BV2 cells.
ESS, extract of Sargassum serratifolium; LPS, lipopolysaccharide; NO, nitric oxide.
Discussion
A wide range of studies have been conducted to investigate the biological activities of natural compounds or extracts from marine macroalgae to develop therapeutic agents due to their easy availability and low side effects. Many brown macroalgae extracts have shown anti-inflammatory activities, moreover, their molecular mechanisms of the extracts from Sargassum sp., such as S. horneri, S. micracanthum, and S. fulvellum, were reported. 14 –16 The present study was designed to investigate the anti-inflammatory mechanism of ESS using LPS-stimulated BV2 microglial cells. Results indicated that ESS at low concentrations efficiently inhibited the secretion of proinflammatory cytokines and inflammatory mediators via the suppression of NF-κB pathway through inhibiting JNK and p38 MAPK phosphorylation in LPS-stimulated BV-2 cells. The inhibitory effect of ESS on the expression of inflammatory mediators suggested one of the mechanisms responsible for its anti-inflammatory action and its potential for use as a therapeutic agent for treating neuroinflammatory diseases.
Physiological levels of NO play a critical role in maintaining normal cell functions, including immune response, neurotransmission, and regulation of blood pressure. However, excess NO and resulting products, as well as reactive nitrogen species (RNS), can induce oxidative damages, eventually cause cell death. 17 NO and RNS produced by activated microglia have been involved in the pathogenesis of neurodegenerative disorders. 18,19 COX-2 is responsible for the synthesis of PGs from arachidonic acid and these PGs activate microglia. Among them, PGE2 is primarily synthesized by microglia in the brain and closely related to neuroinflammation. Excessive levels NO and PGE2 produced by enhanced iNOS and COX-2 are associated with various types of diseases in the central nervous system. The enhanced expressions of iNOS and COX-2 have been observed in microglia in LPS-treated BV2 cells, indicating the important function of NO and PGE2 in neurotoxicity. 20 –22 In this study, we demonstrated that ESS has the ability to inhibit LPS-induced NO and PGE2 production. Moreover, we demonstrated that ESS-mediated inhibition of NO and PGE2 production was due to the suppression of both protein and mRNA levels of iNOS and COX-2 in LPS-stimulated BV2 cells. Interestingly, we found that inhibition of PGE2 by ESS was more marked than that of COX-2 protein and mRNA, and the discrepancy between PGE2 and COX-2 level was caused by the inhibitory activity of ESS on COX-2 enzyme. Thus, the present findings may address that ESS has protective effects on LPS-induced inflammation by its dual functions, transcriptional downexpression of iNOS and COX, and COX-2 inhibition.
Inflammation is further regulated by enhanced levels of proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, produced by activated microglia. 23 Elevated levels of proinflammatory cytokines can stimulate other glial cells to activate, thereby propagating the glial response and consequently the glial-related injury to neurons. 21 Thus, enhanced proinflammatory cytokines are associated with neuropathic diseases, including AD, PD, and multiple sclerosis. 3,5,21 We herein demonstrated the ability of ESS to inhibit production of proinflammatory cytokines in LPS-activated BV2 cells. Moreover, we demonstrated that ESS (20 mg/kg) pretreatment for 4 days suppressed the production of TNF-α and IL-1β in mouse hippocampus. Thus, the present findings may further support the potential to alleviate neuroinflammation.
The expression of iNOS and COX-2 is primarily regulated by transcription factor NF-κB, which is an inactive form bound to IκB in the cytoplasm. 24,25 LPS stimulation induces IκB phosphorylation by IKK, and consequently, IκB is degraded by proteasome after ubiquitination. Thus, the free NF-κB/p65 subunit is translocated into the nucleus. 26 Translocated NF-κB triggers transcription of iNOS and COX-2 as well as proinflammatory cytokines. 27 –29 In this study, we demonstrated that ESS inhibited NF-κB promoter activity by degrading IκB as well as translocating NF-κB/p65 subunit into the nucleus in LPS-stimulated BV2 cells, indicating that NF-κB activation is a major target for anti-inflammatory action of ESS. These results suggest that the ESS-mediated downexpression of iNOS and COX-2 is due to the inhibition of NF-κB activation.
MAPKs (JNKs, p38 MAPK, and ERKs) are involved in the expressions of proinflammatory genes by regulating NF-κB activation. 7 Thus, anti-inflammatory mechanisms are closely associated with regulation of MAPK phosphorylation. Numerous reports have demonstrated that phosphorylation of MAPKs is alternatively necessary for the regulation of NF-κB activation in LPS-stimulated BV2 cells. 20,30,31 In this study, we have demonstrated that ESS inhibits phosphorylation of JNKs and p38 MAPK, but not ERKs, in LPS-stimulated BV2 cells, indicating that JNKs and p38 MAPK are additional targets of ESS. Moreover, ESS showed stronger inhibitory effects on NO production than both inhibitors, which is caused by accumulative action of ESS by inhibiting phosphorylation of JNKs and p38 MAPK. Thus, ESS suppressed LPS-induced inflammatory response by inhibiting the JNK and p38 MAPK pathways as well as by suppressing degradation of IκB in the microglial cells.
In conclusion, we first have demonstrated that ESS inhibits the production of proinflammatory cytokines in vitro and in vivo. The anti-inflammatory mechanisms of ESS were presumed to be the blockade of IκB/NF-κB, JNK, and p38 pathways as well as direct inhibitor of COX-2. The active compounds in ESS were identified as sargahydroquinoic acid, sargachromenol, and sargaquinoic acid. Verification of ESS anti-inflammatory action at the cellular level will be beneficial for further application of ESS in therapeutic agents for neuroinflammation. In vivo studies with isolated compounds will be necessary for the therapeutic applications of this preparation.
Footnotes
Acknowledgment
This research was supported by the Pukyong National University (2015).
Author Disclosure Statement
No competing financial interests exist.