
Abstract
Abnormal activation of adipogenesis in mesenchymal stem cells (MSCs) and preadipocyte cells is associated with human metabolic disorders, such as osteoporosis and obesity. This study investigated the biological effects of protocatechuic acid (PCA) on the modulation of osteogenesis and adipogenesis in cultured cells. PCA stimulation of MSCs significantly increased intracellular mineralization during osteogenesis, but reduced lipid accumulation in both MSCs and 3T3-L1 preadipocyte cells during adipogenesis. Reverse transcription–polymerase chain reaction and immunoblotting analyses showed a dose-dependent upregulation of proosteogenic runt-related transcription factor 2 due to induction of β-catenin. PCA reduced the expression of proadipogenic transcription factor, peroxisome proliferator-activated receptor-γ, and suppressed its promotor activity. These results suggest PCA exerts stimulatory effects on the osteogenesis of MSCs and inhibitory effects on the adipogenesis of MSCs and 3T3-L1 cells. PCA may contribute to maintain a coordinated metabolic balance between adipogenesis and osteogenesis, and thus may be useful for the prevention and alleviation of osteoporosis and obesity.
Introduction
O
Various obesity treatments have been suggested, including inhibition of dietary fat digestion and control of appetite. However, the development of safe and highly effective antiobesity drugs has not been successful and antiobesity drugs are often accompanied by severe side effects, including an increased risk of cardiovascular mortality. 4 Therefore, it is necessary to investigate novel, alternative approaches for obesity treatment. There is growing evidence that nutritional intervention with phytochemicals is effective for controlling obesity and bone loss simultaneously. 5 For example, mesenchymal stem cells (MSCs), derived from bone marrow, are pluripotent stem cells that can be differentiated into several cell types, including osteoblasts and adipocytes. The differentiation of MSCs into adipocytes and osteoblasts is reciprocally regulated; so stimulation of adipogenesis of MSCs could suppress osteogenesis, resulting in osteoporosis. It has been demonstrated that factors activating adipogenesis of MSCs have similar effects on preadipocytes; therefore, the activation of global adipogenesis could increase the risk of developing obesity and could affect osteoblast development simultaneously. There is evidence suggesting that obesity is an independent risk factor for the development of osteoporosis.
Signaling cascades that promote MSC osteogenic and/or adipogenic lineage differentiation generally converge after reciprocal activation of two key transcription factors, peroxisome proliferator-activated receptor-γ (PPARγ) and runt-related transcription factor 2 (RUNX2). Thus, the activation of either transcription factor in MSCs could determine their cellular commitment. Activation of PPARγ along with the CCAAT/enhancer binding protein family is the key to induce a network of signature adipogenic gene expression profiles. A similar gene expression program stimulates adipogenesis of MSCs and preadipocytes. Induction of RUNX2 causes osteogenic target gene expression including OSTERIX.
In addition, it is known that the Wnt signaling pathway plays a key role in controlling the balance between adipogenesis and osteogenesis of MSCs. Activation of β-catenin by stimulation of Wnt signaling plays a critical role in the differentiation of MSCs into mature osteoblasts and, consequently, in bone formation by suppressing adipogenesis. 6 Therefore, the study of these cell commitments has become an area of intense research in recent years since a disruption in the balance between normal osteogenesis and adipogenesis of MSCs has been associated with metabolic disorders such as osteoporosis and obesity.
Phenolic acids are aromatic secondary plant metabolites that are abundantly present in plants. Protocatechuic acid (PCA), a catechol-type O-diphenol phenolic acid (3,4-dihydroxybenzoic acid), has elicited the interest of researchers in the last few years because of its antioxidant activity and additional bioactivities. 7 It has been suggested that phenolic acids have the ability to stimulate bone mineralization 8 ; thus, their supplementation, for example, with PCA, might prevent bone loss induced in animal models. 9 Therefore, in this study, we investigated the effects of PCA on the control of osteogenesis and adipogenesis in a cell culture system.
Materials and Methods
Reagents and cell culture
All reagents were purchased from Sigma-Aldrich unless otherwise stated. Murine C3H10T1/2 MSCs and 3T3-L1 preadipocytes were purchased from Korean Cell Line Bank. C3H10T1/2 MSCs were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM; Hyclone) with 10% heat-inactivated fetal bovine serum (FBS; Hyclone) and 1% penicillin/streptomycin (Hyclone); 3T3-L1 cells were cultured in DMEM with 10% bovine calf serum (Gibco). Cells were cultured in a humidified incubator (37°C, 5% CO2). C3H10T1/2 cells were induced toward osteogenesis at subconfluency following the general protocol described previously
10
by the addition of 50 μg/mL
For adipogenesis, C3H10T1/2 cells were cultured in DMEM (high glucose) containing 50 μg/mL insulin and 1 μM rosiglitazone (Cayman Chemical) for 6 and 9 days. The 3T3-L1 preadipocytes were differentiated for 4 and 8 days by the addition of 10 μg/mL insulin, 0.5 mM 3-isobutyl-1-methyl-xanthine, and 1 μM dexamethasone. To assess the effect of PCA on differentiation, 40 or 80 μM PCA was added to the experimental groups. Differentiation media were replaced every 2 days.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assay
The viability of C3H10T1/2 and 3T3-L1 cells was measured using the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described. 11,12 Cells were cultured on 96-well plates at a density of 1 × 104 and 5 × 104 cells/mL, respectively. Cells were incubated with varying concentrations of PCA (20, 40, 60, 80 and 100 μM of PCA) for 24, 48, and 96 h, followed by the treatment; the culture medium containing 10% of MTT solution (Sigma Aldrich) was added to each well and incubated for 3 h at 37°C. The MTT solution was removed, and the cells were dried for 1 min. Then, 200 μL of dimethyl sulfoxide (DMSO; Bio Basic, Inc.) was added to each well to dissolve the formazan crystals. After mixing DMSO and formazan crystals, the absorbance was measured at 570 nm using a microplate spectrophotometer (Bio-Rad). All experiments were performed in quadruplicate.
Intracellular calcium and lipid staining
Calcium deposition was measured in C3H10T1/2 cells after osteoblastic differentiation for 9 days with or without treatment with PCA. The differentiated osteoblasts were fixed with 70% (v/v) ethanol (Merck Millipore) for 10 min at room temperature, incubated with 2% Alizarin Red S (Sigma-Aldrich) solution (pH 4.2), washed, dried, and microscopically examined (Eclipse T i-s; Nikon). Intracellular calcium, which was stained red, was quantified by both microscopy and spectrophotometry. For image analysis, the calcium-stained area was quantified using image analysis software. 13 For colorimetric analysis, cell lysates were redissolved with 10% cetylpyridinium chloride monohydrate, and the absorbance was measured at 540 nm in a Model 680 microplate reader (Bio-Rad).
Intracellular lipids were analyzed in differentiated adipocytes of C3H10T1/2 and 3T3-L1 cells after 9 and 8 days of differentiation, respectively, by staining with Oil red O (Sigma Aldrich) following the general protocol described previously. 14 After washing with phosphate-buffered saline (PBS), cells were fixed with 10% buffered formaldehyde (Yakuri Pure Chemical) for 1 h at 4°C, incubated with 60% isopropanol for 10 min at room temperature, and stained with Oil red O solution (0.3% oil red O in 60% isopropanol, filtered) for 1 h at room temperature. Intracellular lipids were analyzed by both microscopy and spectrophotometry.
For microscopic analysis, images of cells were obtained using an inverted microscope (Eclipse T i-s; Nikon), and the lipid stained area was quantified using image analysis software. 13 For spectrophotometry, the cell lysates were extracted with 100% isopropanol (Merck Millipore), and the absorbance of samples was measured at 510 nm with a microplate reader (Bio-Rad).
Alkaline phosphatase assay
The alkaline phosphatase (ALP) activity was quantified in C3H10T1/2 cells during osteogenesis by both image analysis and spectrophotometry. ALP expressed in the osteoblasts was determined using the tartrate-resistant acid phosphatase (TRACP) and ALP double-stain kit (Takara) according to the manufacturer's protocol. C3H10T1/2 cells were differentiated into osteoblasts for 6 and 9 days with 40 or 80 μM of PCA, and cells were fixed with citrate buffer (pH 5.4) containing 60% acetone and 10% methanol for 5 min at room temperature. Then, cells were incubated with bromo-chloro-indolyl phosphate/nitro blue tetrazolium chloride substrate for ALP for 45 min at 37°C. After incubation, the solution was removed and the cells were washed thrice with sterilized distilled water. For microscopic analysis, cells were imaged using an inverted microscope (Eclipse T i-s; Nikon). The stained area of the cells was measured using Image software analyzer. 13 Spectrophotometric analysis was measured using the TRACP and ALP assay kit (Takara) as described in the kit protocol with some modifications. Osteoblasts were washed with 0.85% physiological saline buffer, lysed with acetone solution (acetone:water = 8:2, v/v), and incubated with ALP substrate solution (12.5 mM p-nitro-phenyl phosphate) at 37°C for 60 min. The reaction was stopped with NaOH solution (0.9 N) and the absorbance measured at 405 nm using a microplate reader (Model 680; Bio-Rad). The measured absorbance values were normalized by protein concentration and quantified using the Bradford assay 15 with bovine serum albumin (BSA; Bavogen) as the standard.
Measurement of fatty acid oxidation and synthesis rates
The rate of fatty acid oxidation was quantified following the general protocol described previously. 10 On day 8 or 9 of adipogenic differentiation, cells were lipid loaded with 0.44 mM (1- 14 C) palmitate containing 1 mg/mL fatty acid-free BSA in DMEM. After incubation for 1 h in a humidified incubator (37°C, 5% CO2), the cell medium was collected and transferred to counter tubes containing 0.5 mM NaOH (Daejung) and 0.5 mM HCl (Junsei). After vigorous mixing, samples were incubated for 30 min at room temperature. The radioactivity was determined by an LS-6500 liquid scintillation counter (Beckman Instruments).
The rate of fatty acid synthesis was also measured as described previously. 10 Fully differentiated adipocytes were incubated with 0.96 mCi/mole (1- 14 C) acetate in DMEM (Hyclone) for 1 h at 37°C. Cells were washed twice with KCl (0.14 M) and the reaction was stopped with 1.5 mL of NaOH (0.5 M). The reaction solution was saponified with 4 mL of ethanol and 2 mL of distilled water for 90 min at 90°C. After acidification with 1 mL of HCl (7 M), unsaponifiable sterol and fatty acids were extracted with 3 mL of petroleum ether. After drying overnight, radioactivity was measured in the extracts using an LS-6500 liquid scintillation counter, and cell monolayers were recovered to determine the protein concentration. All experiments were performed in quadruplicate.
Intracellular lipid measurements
Fully differentiated adipocytes were washed twice with PBS, and intracellular triglycerides and cholesterol were extracted with a mixture of hexane and isopropanol (2:1 ratio) for 30 min at room temperature. The cell extracts were then moved to tubes, and the solvent was evaporated at 30–40°C using a rotary evaporator (Eyela). The remaining lipids were diluted in 95% ethanol, and their concentration was measured with a Cobas C111 automatic analyzer (Roche Diagnostic Systems, Inc.). Intracellular triglyceride and cholesterol concentrations were normalized using the concentration of cellular protein, as measured with the Bradford assay.
Total RNA isolation and reverse transcription–polymerase chain reaction
Cultured cells were collected by scraping, and RNA was extracted using RNAiso Plus (Takara) according to the manufacturer's protocol. One microliter of RNA was reverse transcribed to cDNA using the ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo). Primer sequences are shown in Table 1. Reverse transcription–polymerase chain reaction (RT-PCR) products were made using the EmeraldAmp GT PCR Master Mix (Takara). The products were electrophoresed on 4% agarose gels containing ethidium bromide according to standard protocols. The relative band densities were normalized to glyceraldehyde 3-phosphate dehydrogenase and analyzed by ImageJ software analyzer. 13
Immunoblottings
Western blot analysis was performed to quantify RUNX2 and PPARγ protein expression. Cells were washed with cold PBS, lysed in RIPA buffer (Thermo Scientific), separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and then electrotransferred to nitrocellulose membranes (0.45 μm pore size; Whatman). After the transfer, each membrane was blocked using a solution of 0.001 g/mL Tween-20 in TBS containing 0.05 g/mL nonfat dry milk for 1 h at room temperature and then incubated with rabbit anti-RUNX2 (Santa Cruz Biotechnology) or mouse anti-PPARγ (Santa Cruz Biotechnology) antibody overnight at 4°C.
Bound primary antibodies were detected using the peroxidase-coupled anti-mouse or anti-rabbit immunoglobulin G secondary antibodies (Thermo Scientific) at 1:5000 and 1:8000 dilutions, respectively, exposed to SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) for protein detection using the ChemiDoc Touch Imaging System (Bio-Rad). Digitally acquired blots were adjusted for brightness and contrast; image resolution was kept constant. Densitometry was performed with ImageJ software 13 and relative RUNX2 and PPARγ band intensity normalized to β-actin and quantified with respect to control.
Reporter gene assay
HEK293 cells were grown in DMEM with 10% FBS and 1% penicillin/streptomycin (Hyclone) at 37°C in a 5% CO2 humidified incubator. Cells were cotransfected with PPRE luciferase reporter, Renilla luciferase vector, and PPARγ expression vectors using lipofectamine reagent (Invitrogen) and incubated for 24 h. Then, cells were treated with 40 or 80 μM PCA or 10 μM troglitazone for an additional 24 h. The luciferase activity was measured using a luciferase assay detection kit (Dual-Luciferase Reporter Assay System; Promega; E1910) according to the manufacturer's protocol, and the luminescence was quantified using a VICTOR™ X Light luminescence late reader (PerkinElmer; 2030-0010). The relative firefly luciferase activity was normalized using the Renilla luciferase activity as arbitrary luciferase units (Firefly/Renilla).
Statistical analysis
Measurements were performed at least in triplicate. All data were analyzed with GraphPad Prism 5 software (GraphPad). Multiple group comparisons were performed using one-way analysis of variance, followed by Tukey's test. Differences at P < .05 were considered to be significant. All data were expressed as the mean ± standard error of the mean.
Results
PCA stimulates mineralization during osteogenesis, while suppressing lipid accumulation during adipogenesis of MSCs and preadipocytes
First, the cytotoxicity of PCA was examined in MSCs and 3T3-L1 cells. PCA (20–100 μM) treatments for 24, 48, and 96 h did not significantly alter cell viability (Fig. 1); thus, 40 and 80 μM PCA were selected for further experiments. To investigate whether PCA modulates the osteogenesis and adipogenesis, the intracellular mineralization and lipid accumulation of C3H10T1/2 and 3T3-L1 cells were assessed. In C3H10T1/2 cells, calcium deposition, assessed by Alizarin red S staining, was significantly increased by PCA (40 μM) on days 6 and 9 of osteogenesis (Fig. 2A). In addition, PCA significantly reduced lipid contents in both C3H10T1/2 and 3T3-L1 adipocytes, assessed by Oil red O staining (Figs. 4A and 5A). The antiadipogenic effect of PCA was greater in 3T3-L1 preadipocytes on days 4 and 8 of adipogenesis than the observed effect in C3H10T1/2 cells. (Fig. 5A). These results suggest that PCA exhibits proosteogenic and antiadipogenic activity.
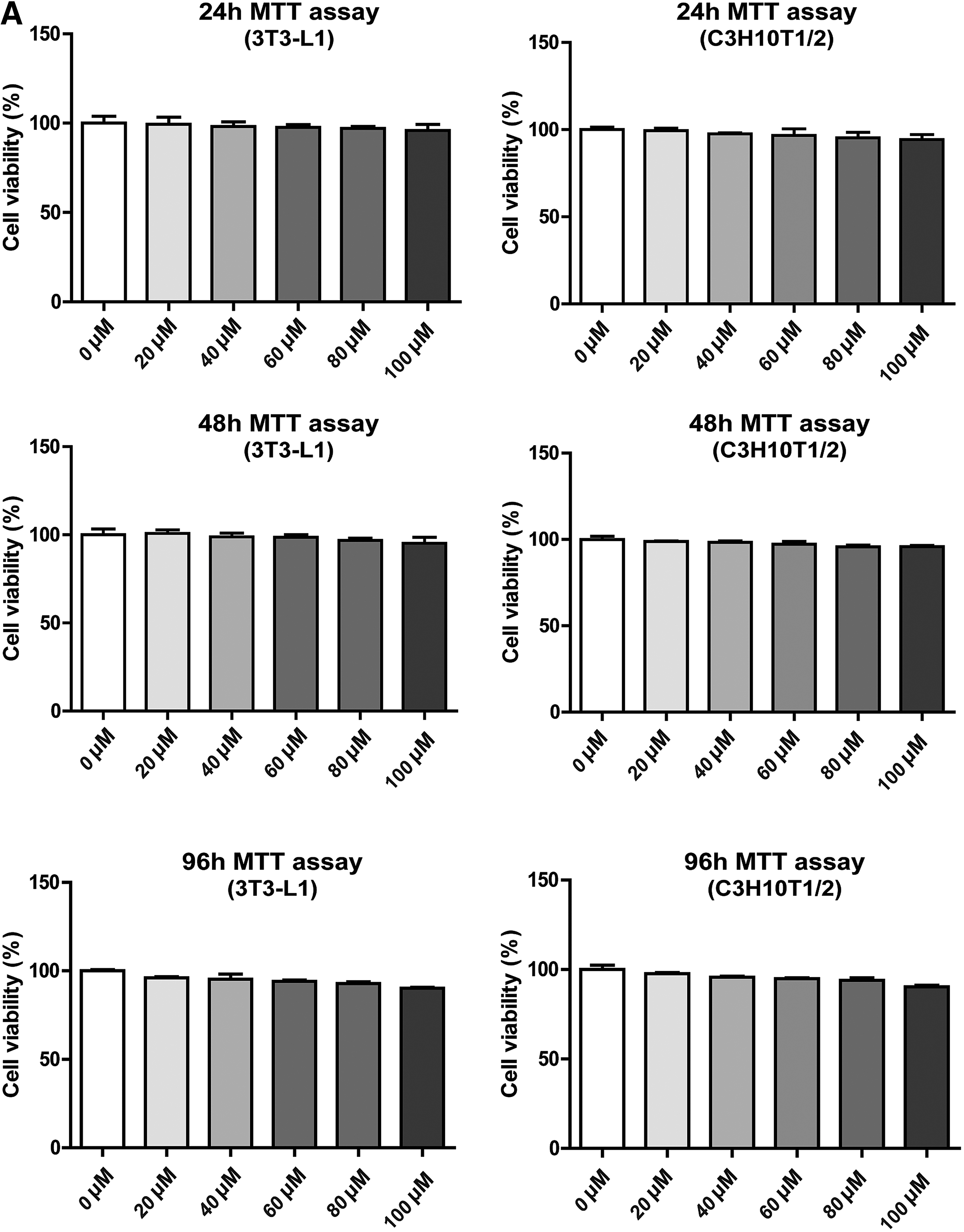
MTT cell viability assay by PCA.
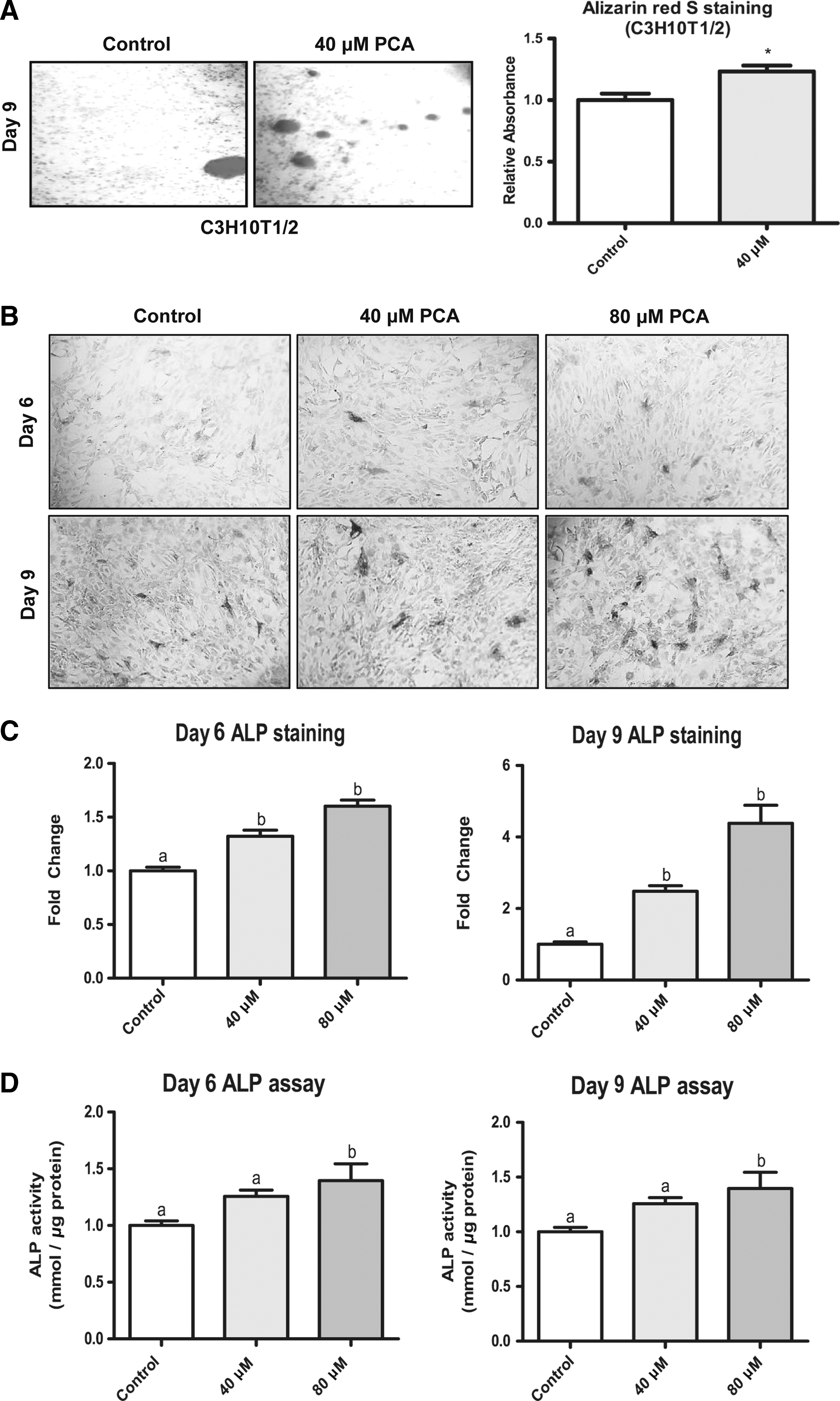
PCA stimulates ALP activity during osteogenesis of C3H10T1/2 MSCs.
PCA stimulates ALP activity during osteogenesis of MSCs
The proosteogenic effects of PCA were further evaluated by the ALP activity assay because ALP expression and activity are hallmarks of osteoblastic induction. The ALP activity was analyzed by both image analysis and spectrophotometry. Image analysis revealed that the ALP activity was increased by PCA after osteogenesis because ALP staining area increased from day 6 to 9 after osteogenic induction (Fig. 2B). Cells stimulated with 80 μM PCA showed a higher level of ALP activity compared to those incubated with only 40 μM PCA. The results of spectrophotometric analysis confirmed the above findings (Fig. 2C). The ALP activity was similar in cells stimulated with 80 μM PCA after 6 days of differentiation, but significantly higher in cells after 9 days (Fig. 2D). These results show that PCA increased the ALP activity, confirming the proosteogenic activity of PCA in MSCs.
PCA induces the expression of genes involved in osteoblast commitment
We next investigated the effect of PCA on osteoblastic gene expression of MSCs by RT-PCR. The results show that the expression of RUNX2, ALP, and OSTERIX was significantly upregulated in MSCs stimulated with PCA (80 μM) on both day 6 and 9 of differentiation (Fig. 3A). The upregulation level of RUNX2, a master osteogenic transcription factor, was maintained between days 6 and 9. Despite PCA being stimulated in a dose-dependent manner, results showed a slight reduction of RUNX2 gene expression in cells treated with 40 μM on day 9. These results are consistent with previous reports that RUNX2 is a key gene expressed in the early stages of the osteogenic differentiation process.
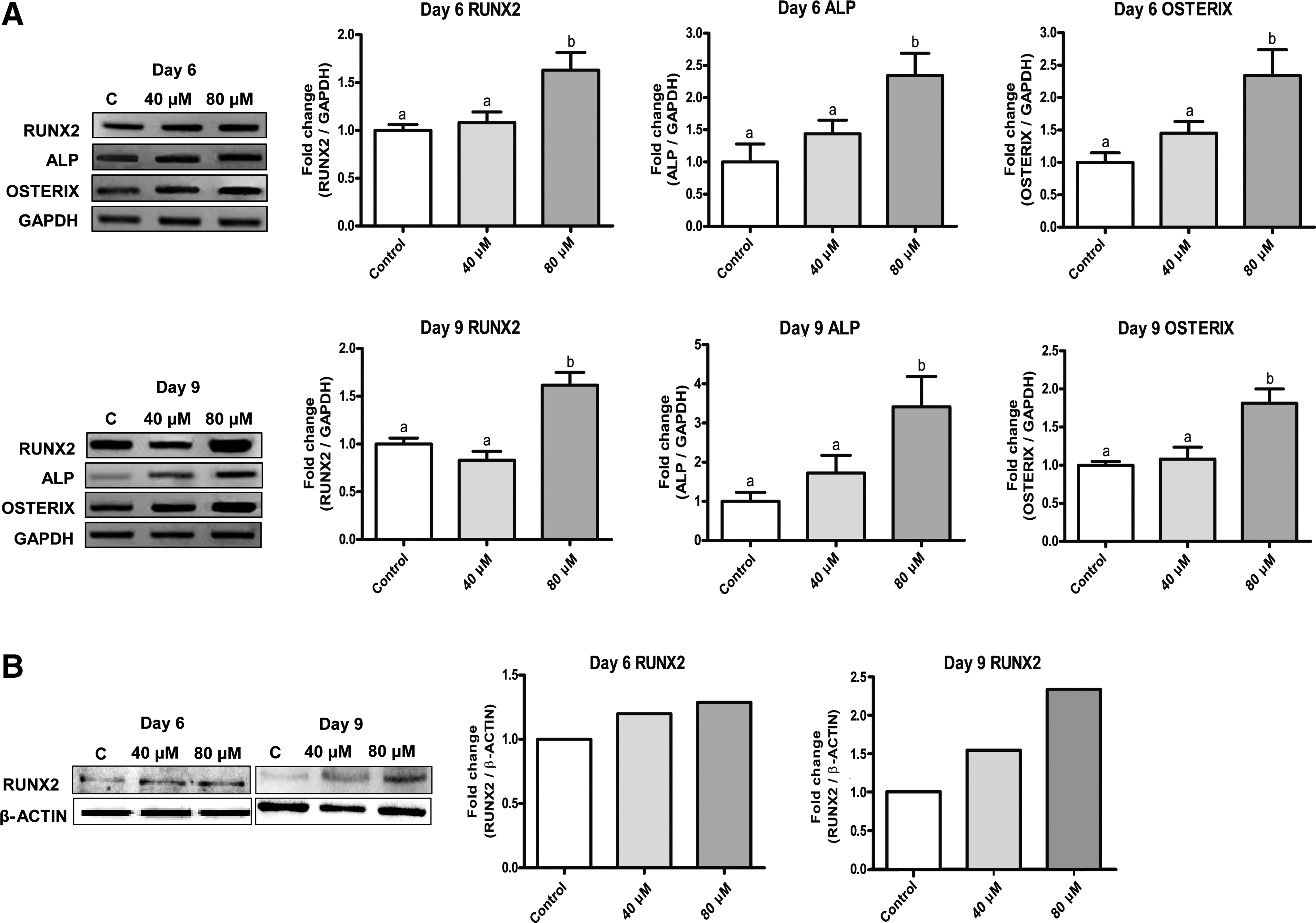
PCA stimulates osteogenic gene expression in C3H10T1/2 MSCs.
Furthermore, ALP expression was induced to a greater extent compared to RUNX2 on both day 6 and 9 (Fig. 3A). PCA at 80 μM induced ALP mRNA expression up to 3.4-fold on day 9 of differentiation. OSTERIX gene expression peaked on day 6, but then slightly declined on day 9 in cells treated with 80 μM PCA (Fig. 3A). PCA at 40 μM showed only minimal effects on the expression of the OSTERIX gene. The protein expression of RUNX2, assessed by Western blot analysis, was induced by 1.2- and 2.3-fold, respectively, with 80 μM PCA on day 6 and 9 of differentiation compared to the control (Fig. 3B). These results suggest that PCA stimulates mRNA and protein expression of RUNX2, indicating its proosteogenic activity.
PCA reduces the rate of fatty acid synthesis, but increases the rate of fatty acid oxidation by suppressing PPARγ protein expression in adipocytes
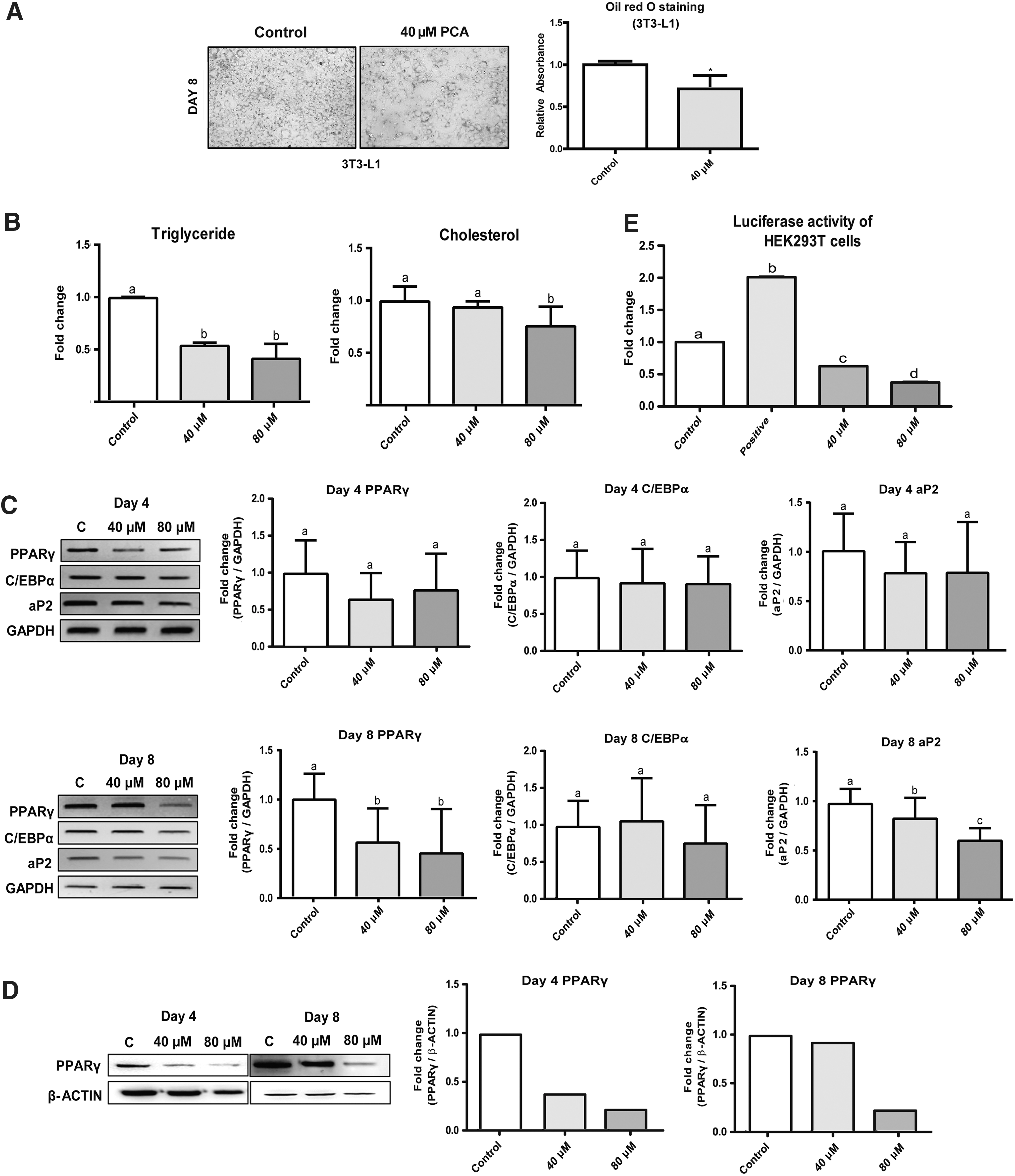
As shown earlier, PCA reduced the lipid accumulation in MSCs and 3T3-L1 cells during adipogenesis (Figs. 4 and 5A, respectively). In line with this finding, PCA reduced intracellular triglyceride and cholesterol concentrations in MSCs as well (Fig. 4B). Quantification of fatty acid metabolism shows that PCA increased the rate of fatty acid oxidation, while decreasing the fatty acid synthesis rate (Fig. 4C). This suggests that the lowered intracellular lipid levels are due to modulation of the fatty acid metabolic rate. The mRNA expression of key adipogenic genes tended to decrease, but this tendency did not reach statistical significance (Fig. 4D); however, the protein expression of PPARγ was dramatically suppressed by 87% with 80 μM PCA (Fig. 4E).

The effects of PCA on the adipogenesis of C3H10T1/2 MSCs.
The effects of PCA on the adipogenesis of 3T3-L1 cells.
PCA also reduced intracellular triglyceride levels in 3T3-L1 adipocytes (Fig. 5B). Similar to the findings from MSCs, PCA tended to suppress adipogenic gene expression. After full differentiation, on day 8, the mRNA expression levels of both PPARγ and aP2 were significantly suppressed due to PCA (Fig. 5C). Immunoblotting analysis shows that the protein level of PPARγ was intensely reduced on day 8 (Fig. 5D). Reporter gene, determined by Luciferase assay, showed that PCA significantly suppressed the PPARγ promotor activity by 1.4-fold and the effect was dose dependent (Fig. 5E). These results collectively suggest that PCA suppresses adipogenesis by reducing PPARγ protein expression levels and modulates fatty acid metabolism rates.
PCA induces the expression of genes in the Wnt/β-catenin signaling pathway
Reciprocal regulation of osteogenesis and adipogenesis in MSCs could be triggered by modulation of the Wnt/β-catenin signaling pathway. Thus, we performed RT-PCR to quantify key gene expressions in the Wnt signaling pathway in cells stimulated with PCA. The mRNA level of Wnt3a tended to be increased on both day 6 and 9, but the level of β-catenin showed a robust 2.4-fold increase in cells on day 6 of stimulation with 80 μM PCA compared to the mRNA level in control cells (Fig. 6). The induction of β-catenin mRNA by PCA was maintained on day 9 of osteogenesis. These results suggest that activation of the Wnt/β-catenin signaling pathway may contribute to the activation of osteogenesis and suppression of adipogenesis in MSCs.
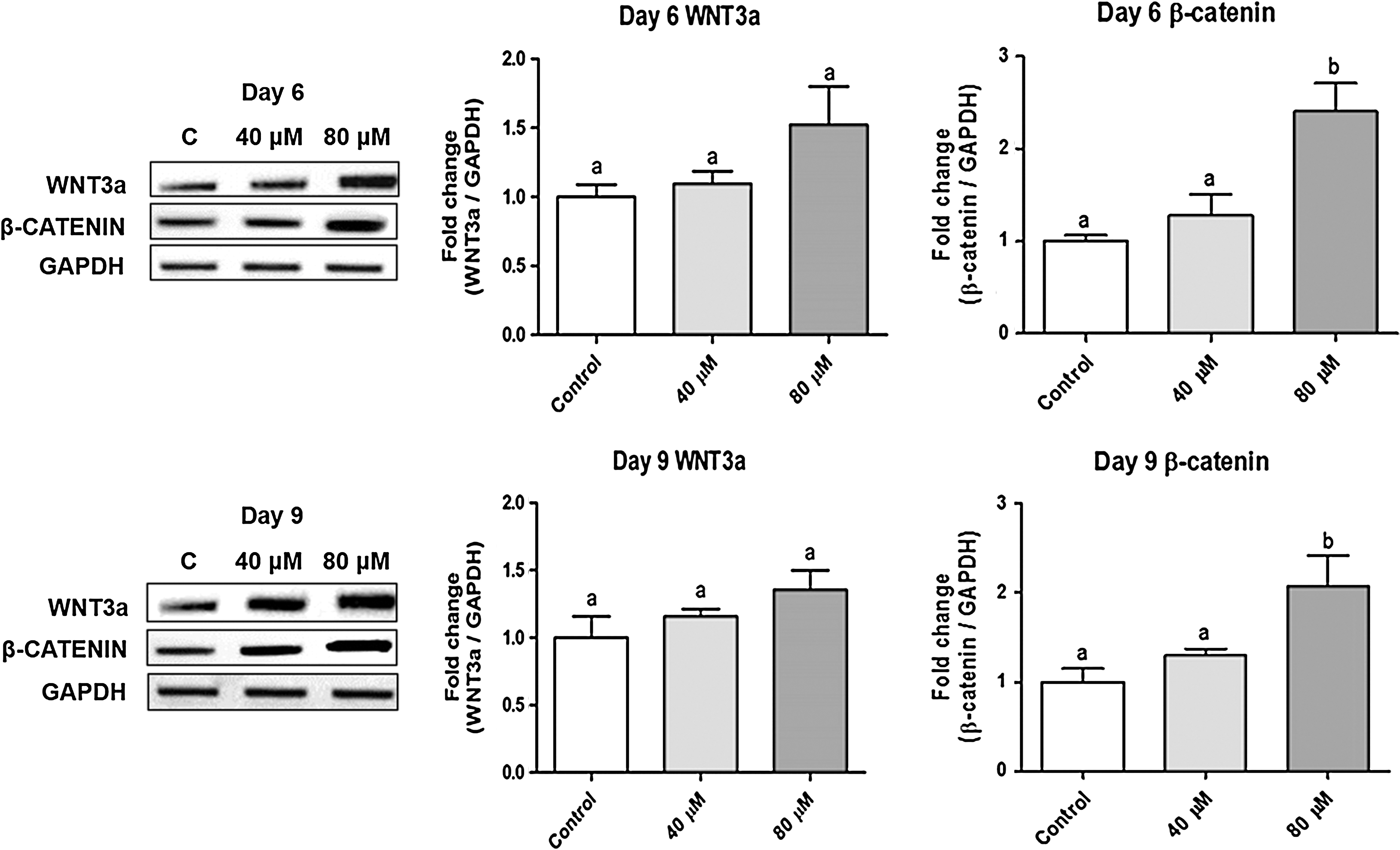
PCA stimulates genes in the Wnt/β-catenin signaling pathway. The mRNA expression of genes was measured by RT-PCR. The density of each band was analyzed by ImageJ software and the relative expression was normalized to the level of glyceraldehyde 3-phosphate dehydrogenase. N = 4–6 per group. Group comparisons were performed using ANOVA followed by Tukey's test with GraphPad Prism software. Values that do not share the same letter are significantly different (P < .05).
Discussion
PCA is a naturally occurring phenolic acid that is thought to have several biological activities, including antioxidative, antidiabetic, antiaging, anticancer anti-inflammatory, and antiobesity effects. 16 –18 The general mechanisms of adipogenic inhibition by phenolic compounds involve their activity as antioxidants, as well as the inhibition of lipid mediator generating enzymes. 7 However, the mechanism of PCA in the prevention of obesity may be much more complex because homeostasis of energy metabolism involves multiple organs. PCA is considered the main human metabolite of C3G and may be responsible for several health benefits ascribed to anthocyanin-rich food consumption.
PCA is a very common compound present in human diet, present in bran and grain brown rice (Oryza sativa L.) and onion (Allium cepa L.), especially in the scales. It is a also detected in many fruits, such as plums (Prunus domestica L.) and grapes (Vitis vinifera). Juice from blood oranges (Citrus aurantium) is another important dietary source of C3G and PCA 19 and has been shown to have a beneficial effect on obesity and glucose homeostasis in mice by improving glucose tolerance and insulin sensitivity. 20 However, it is imperative to mention that there might be unwanted effects on insulin resistance. Further studies should be performed over this matter. Nevertheless, studies have reported an antiulcer activity and antidiabetic and antiseptic properties, in addition to displaying anticoagulant effects in diabetic mice. 21 In humans, PCA in the bloodstream 6 h postconsumption accounts for 44.4% of the ingested cyanidin-3-O glucoside. 19
Furthermore, in vitro experiments have demonstrated that PCA, like other polyphenols, is able to inhibit low-density lipoprotein (LDL) oxidation, and the inhibition of lipid mediator generating enzymes is one of the PCA anti-inflammatory mechanisms, 7,22 and the possibility of using PCA as a hypoglycemic agent and in other human applications has arisen 23 Moreover, in streptozotocin-diabetic mice, it has been demonstrated that PCA improves glycemic control and attenuates homeostatic disorder, hence having an insulin-like activity by increasing the glucose uptake and enhancing the glucose transporter 4 translocation and adiponectin secretion in human primary adipocytes. 7 Thus, it has been suggested that the biological activities of anthocyanins may be explained at least, in part, by PCA. 17,23
Obesity is characterized by an increase in adipose tissue mass. An increase in both the size and number of adipocytes is critical for fat accumulation. Although dietary intervention and physical exercise can control obesity, inhibition of adipogenesis and fat accumulation is an alternative strategy for obesity therapy. PCA may be a candidate for this purpose because it suppressed adipogenesis of 3T3-L1 cells. In addition, we examined the mechanism of PCA on the commitment of C3H10T1/2 MSCs to the osteogenic lineage rather than the adipogenic lineage, and our results suggest that PCA promoted osteogenesis through activation of the key osteogenic transcription factor RUNX2. Adipogenesis of MSCs, however, was suppressed by PCA by a reduction in gene and protein expression of the master adipogenic transcription factor PPARγ.
To our knowledge, this is the first report to show that PCA is a modulator of MSC and preadipocyte differentiation by inducing osteogenesis and inhibiting adipogenesis. PCA stimulated the osteogenic marker ALP activity and osteogenic calcium deposition in MSCs. Simultaneously, PCA reduced lipid accumulation and adipogenesis in both MSCs and preadipocytes. These effects were shown at noncytotoxic concentrations of PCA. Our results suggest that PCA-stimulated osteogenesis may be due to controlling the lineage-specific transcriptional program on the basis of upregulating RUNX2 and suppressing PPARγ gene expression. RUNX2 is an early and key transcription factor, initiating the osteogenic lineage transcriptional program that results in the upregulation of bone formation genes such as OSTERIX, a late bone formation marker that is required to form bone matrix. In addition, the suppression of PPARγ gene expression by PCA may contribute to RUNX2-mediated osteogenic transcription indirectly because it is known that PPARγ has an antagonistic effect on osteogenic cellular signaling factors such as transforming growth factor β (Fig. 7). However, the exact biological mechanism of the interaction of RUNX2 and PPARγ needs further future investigation.
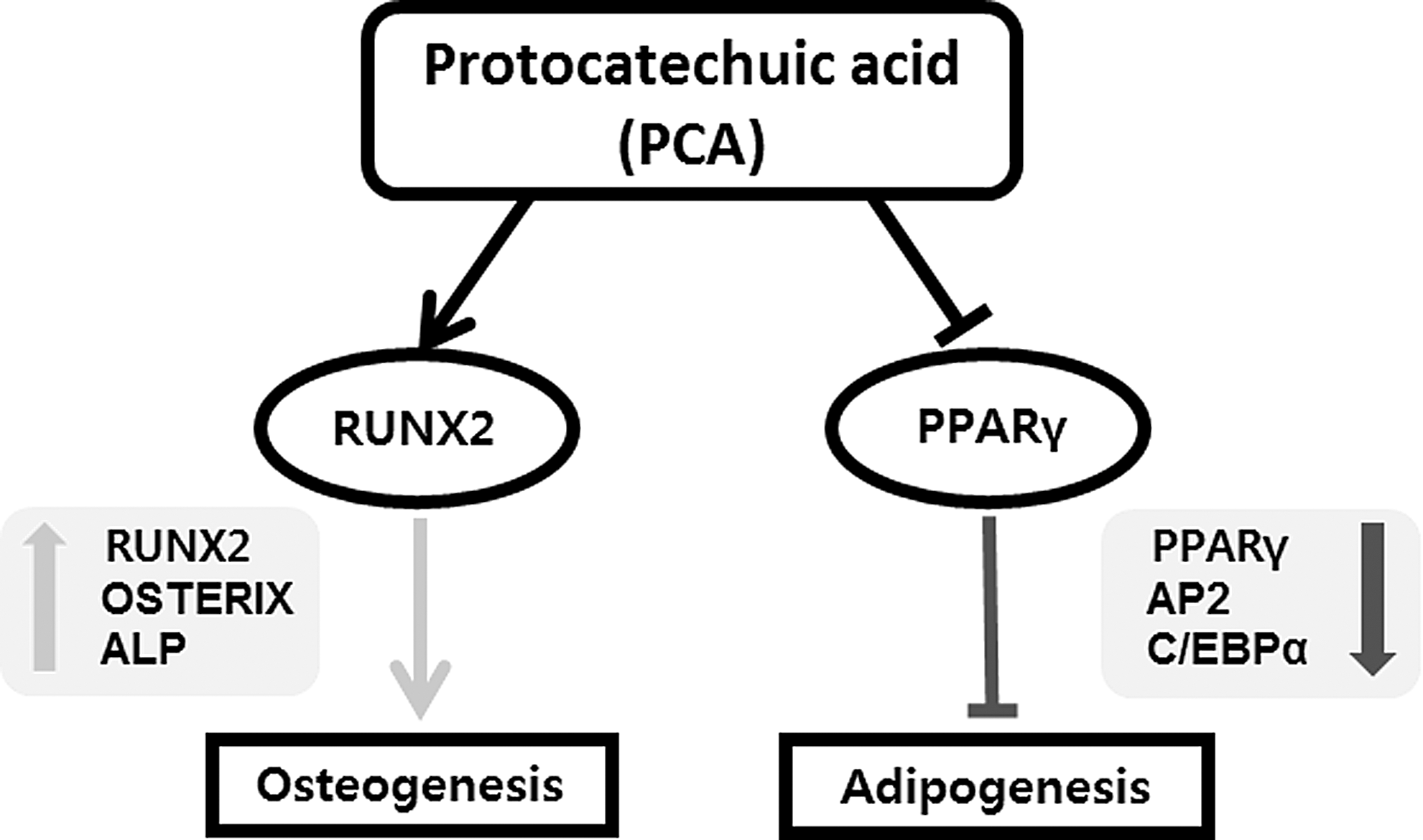
The summary of biological effects of PCA on the modulation of osteogenesis and adipogenesis. PCA activates osteogenesis, but suppresses adipogenesis by reciprocal regulation of RUNX2 and PPARγ. Induction of RUNX2 stimulates the expression of osteogenic genes, including OSTERIX and ALP, and the suppression of PPARγ reduces expression of adipogenic genes such as aP2 and C/EBPα. These regulatory effects of PCA may contribute to the prevention of osteoporosis and obesity simultaneously.
The results also suggest that PCA promotes the Wnt/β-catenin signaling pathway, thereby inducing RUNX2 expression. In MSCs, PCA stimulated β-catenin gene expression in a dose-dependent manner, which induces osteogenic gene programming of MSCs (Fig. 7). Canonical Wnt signaling is an important osteogenesis-controlling pathway. 24 However, the role of Wnt signaling in MSC osteogenic differentiation is complex and depends on the cellular context. It is known that dysregulation/hyperactivation of Wnt signaling is associated with numerous diseases, including osteoporosis. To date, over 19 Wnt receptors and co-receptors have been identified within seven families of proteins. In general, Wnt signaling has demonstrated both proosteogenic and antiosteogenic activities through both canonical and noncanonical pathways. The β-catenin-dependent canonical pathway starts with the binding of extracellular Wnt ligands to the frizzled receptors expressed at the cell surface. This induces complex formation with the LDL (LRP5/6) coreceptors, as well as with the intracellular proteins of the disheveled family. The resulting activation of disheveled protein then functions to inhibit a second, intracellular complex comprising Axin, GSK3β, and adenomatous polyposis coli (APC) proteins. GSK3β normally phosphorylates β-catenin, promoting its degradation. Wnt stimulation inhibits the Axin/GSK3β/APC complex, and β-catenin accumulates instead of being degraded, leading to an increase in nuclear β-catenin levels. Once inside the nucleus, β-catenin can heterodimerize with a member of the lymphoid enhancer factor/T cell factor family. Ultimately, canonical Wnt signaling elicits gene transcription activity that influences MSC lineage determination.
In our results, the expression level of Wnt3a, one of the major Wnt factors expressed in MSCs, was not significantly altered by PCA, but the mRNA expression of β-catenin was significantly increased depending on PCA dose. Therefore, PCA may activate the Wnt/β-catenin signaling pathway by inducing β-catenin expression at least in part. This induction may contribute to the upregulation of RUNX2 and subsequent activation of the osteogenic gene expression program in MSCs.
In this study, stimulation of PPARγ and RUNX2 by PCA exposure during adipogenesis and osteogenesis was investigated in C3H and 3T3-L1 cells. PCA had significant effects, enhancing osteogenic and inhibiting adipogenic differentiation by stimulating canonical Wnt signaling in C3H10T1/2 cells. This suggests that PCA is a new potent antiadipogenic and proosteogenic agent. In addition, PCA deserves great nutritional interest as the main human metabolite of C3G, which in turn is the most representative dietary anthocyanin. 20 However, additional studies on the metabolic effects of PCA are warranted to elucidate the exact mechanism(s) of adipogenic inhibition.
Footnotes
Acknowledgment
This work was carried out with the support of the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF-2016R1A2A2A05005005483), and a grant (PJ01125304) from Cooperative Research Program for Agriculture Science & Technology Development, Rural Development Administration, Republic of Korea.
Author Disclosure Statement
No competing financial interests exist.