
Abstract
Nuclear factor E2-related factor 2 (Nrf2) is the master regulator of antioxidant enzymes and is known to act on the nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) signaling pathway. Few studies have examined the bioactivity of halleridone. Herein, we investigated whether halleridone, which was isolated from the stems of the plant Cornus walteri, could regulate Nrf2-mediated heme oxygenase (HO)-1 expression and prevent intramicroglial inflammation induced by amyloid beta (Aβ)1-42 overexpression. Biochemical and molecular experiments, such as real-time polymerase chain reaction, Western blot analysis, immunocytochemistry, immunofluorescence, and luciferase reporter gene assays, were performed. The results demonstrated that halleridone promoted the upregulation of Nrf2 expression and its translocation to the nucleus, thereby activating antioxidant response element gene transcription and HO-1 expression in murine hippocampal HT22 cells. Additionally, halleridone removed intramicroglial Aβ 1-42 and suppressed the production of inflammatory mediators such as interleukin (IL)-1β, IL-6, prostaglandin E2, and nitric oxide (NO) induced by artificially overexpressed Aβ 1-42 and decreased pNF-κB accumulation in the nucleus and the expression of inducible NO synthase and cyclooxygenase II in BV-2 cells. In conclusion, halleridone activated Nrf2-mediated HO-1 expression and inhibited Aβ 1-42-overexpressed microglial BV-2 cell activation. These observations suggest that halleridone may have therapeutic potential for targeting neurodegeneration through neuroinflammation.
Introduction
A
Nuclear factor E2-related factor 2 (Nrf2) is a known master regulator of antioxidant responses 4 and is an upstream transcription factor that induces the expression of antioxidant enzymes, including heme oxygenase (HO)-1 (EC 1.14.14.18). It is known to exhibit strong antioxidant and cytoprotective effects during heme degradation to generate carbon monoxide, ferritin, biliverdin, and bilirubin. 5,6 The Nrf2/Kelch-like ECH-associated protein 1 (Keap1)-mediated HO-1 pathway is regulated by phosphorylation of mitogen-activated protein kinase/Akt 7,8 or phosphatidylinositol-3-kinase (PI3K)/Akt. 9
Several studies have reported that Nrf2 controls inflammatory response and that signal activation of the Nrf2-antioxidant enzyme can attenuate the nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB)-mediated inflammatory response. 6,10 Inflammatory mediators produced by NF-κB-mediated gene regulation induce phagocytic and cytotoxic activities and eventually macrophage activation. Phosphorylated NF-κB translocates to the nucleus through external stimulation or the cytokine activation pathway, and phosphorylated NF-κB is regulated by IκB kinase activation. 11,12
Halleridone was isolated from Halleria lucida (Scrophulariaceae), 13 Teucrium decipiens, 14 and Phyla nodiflora. 15 Its bioactivity and therapeutic approach to antioxidant and antineuroinflammatory activities are poorly understood. Thus, we investigated whether halleridone has HO-1-inducing activity in hippocampal HT22 cells and NF-κB-dependent inflammation inhibitory activity in microglial BV-2 cells.
Materials and Methods
Cell culture
Immortalized murine hippocampal HT22 and human neuroblastoma SH-SY5Y cell lines were kindly provided by Prof. Kyung-Sik Song and Dong-Seok Lee from Kyungpook National University. A murine microglial macrophage BV-2 cell line was provided by the Korea Research Institute of Bioscience and Biotechnology (KRIBB). All cell lines were grown in Dulbecco's modified Eagle's medium (DMEM)/high-glucose medium supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin and 100 μg/mL streptomycin (P/S).
Chemicals and reagents
Halleridone was provided by the Korea Bioactive Natural Material Bank (KBNMB) and its structure is presented in Figure 1A. All reagents for cell maintenance were provided by HyClone (GE Healthcare, United Kingdom). Antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA), Abcam (Cambridge, MA, USA), and Cayman (Ann Arbor, MI, USA). Most chemicals and reagents used for molecular work were of high-quality grade.
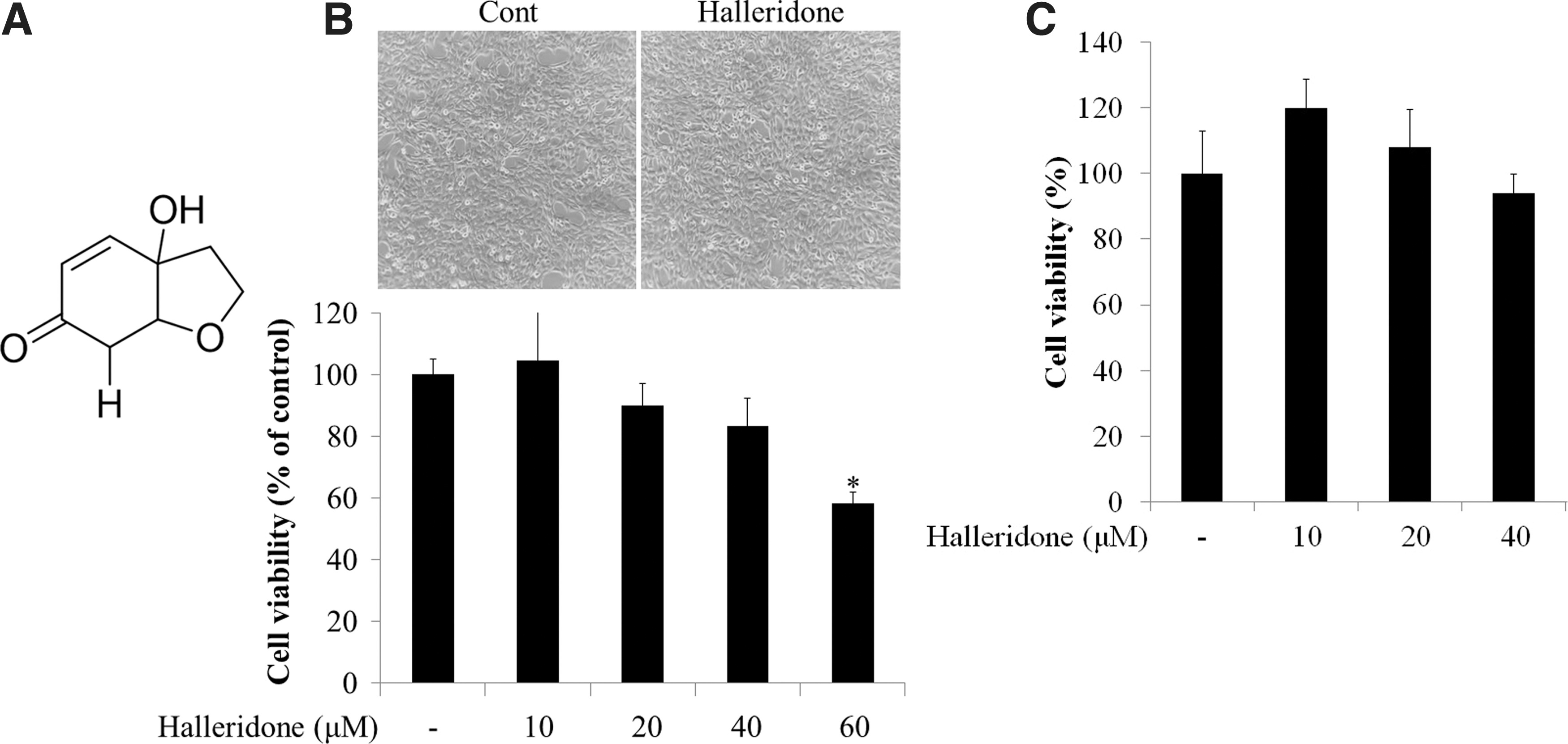
The structure of halleridone and its cytotoxicity.
Real-time polymerase chain reaction analysis
The experiment was performed following previous protocols with minor modification. 16 In this study, 300 μg of synthesized cDNA was subjected to a StepOne Real-Time polymerase chain reaction (PCR) System.
Western blot analysis
Western blot analysis was performed by following previous protocols. 16 Briefly, sodium dodecyl sulfate–polyacrylamide gel electrophoresis was performed using an electrophoresis kit obtained from Bio-Rad (Hercules, CA, USA). A sample of 30 μg of protein was loaded onto a gel at 50 V for 30 min and 200 V for 1–2 h. The proteins on the gel were transferred to Immobilon®-P 0.45 μm polyvinylidene difluoride transfer membranes purchased from Millipore (Billerica, MA, USA). The membranes were soaked in 5% skim milk solution for 1 h at room temperature or overnight at 4°C. Then, the membranes were rinsed twice with 0.2% Tween 20® dissolved in Tris-buffered saline.
Membranes were treated with primary antibodies and incubated for three and half hours at room temperature or overnight at 4°C. The membranes were washed and treated with horseradish peroxidase-conjugated anti-rabbit, anti-goat, or anti-mouse-immunoglobulin G secondary antibodies and incubated for 2 h at room temperature. The membranes were washed again and treated with chemiluminescent substrates from Thermo Scientific (Rockford, IL, USA). The target proteins were visualized using Image Quant™ LAS4000 from GE Healthcare Life Sciences (Little Chalfont, United Kingdom). Relative protein expression levels were calculated using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Antioxidant response element reporter gene assay
A stable HT22-antioxidant response element (ARE) cell line transfected with a pGL4.37[luc2P/ARE/Hyg] vector (Promega Corp., Madison, WI, USA) from our previous study 17 was used in this assay. Briefly, cells were seeded at 3 × 105 cells per well in a 12-well plate. The cells were starved by growing in DMEM supplemented with 1% FBS and P/S for 12 h and were then treated with each sample for 16 h. Cells were lysed by treating with 120 μL of passive lysis buffer for 30 min with vigorous agitation every 10 min. Cell lysates were isolated by centrifugation at 12,000 g for 2 min. Luminescence was measured in the collected supernatants using a SpectraMax L microplate reader (Molecular Devices, San Francisco, CA, USA). ARE-luciferase activity was expressed as relative induction compared with controls.
Preparation of Aβ 1-42 construct and transfection
The plasmids for overexpressing Aβ 1-42 were constructed with enhanced green fluorescent protein (EGFP) and kanamycin resistance genes. The construct map is shown in Figure 4A. Cells were transfected with Aβ1-42 plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 12 h following manufacturer's protocols.
Immunocytochemistry and immunofluorescence
HT22 cells (3 × 105 cells/plate) were treated with halleridone in medium containing 1% FBS and P/S for 12 h and were subsequently fixed with 3.7% formalin solution for a week. Membrane permeabilization was carried out using 0.2% Triton X-100 dissolved in phosphate-buffered saline (PBS), and the cells were blocked with 1% bovine serum albumin solution. The cells were treated with primary antibodies at a 1:200 ratio overnight at 4°C, following the provider protocols. After discarding the solution with primary antibodies and washing with PBS five times, the fixed cells were treated with secondary antibodies, including anti-rabbit fluorescein isothiocyanate (FITC) and anti-goat Texas red (TR), at a 1:500 ratio for 2 h. Subsequently, the cells were stained with 1 μg/mL of 4′,6-diamidino-2-phenylindole (DAPI) solution for 1 min.
Cells were then washed with PBS five times and mounted using Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). Confocal immunofluorescence images were taken using a TCS SP8 confocal microscope (Leica, Wetzlar, Germany). For immunofluorescence imaging, BV-2 cells were transfected with Aβ 1-42 plasmids using Lipofectamine 2000 and with halleridone for 12 h. Fluorescence images were taken using fluorescence microscopy (Olympus, IX70, Japan).
Cytokine, prostaglandin E2, and nitric oxide assays
The secreted amounts of interleukin (IL)-6, IL-1β, or prostaglandin E2 (PGE2) from BV-2 cells were separately measured using mouse IL-6 (BD Biosciences, San Jose, CA, USA), mouse IL-1β (Thermo Scientific), and PGE2 (Thermo Scientific) ELISA kits according to manufacturer protocols with slight modifications. The Griess method was used for nitric oxide (NO) assays. 18 To measure NO production, absorbance was detected at 540 nm wavelength using a VersaMax™ microplate reader (Biocompare, San Francisco, CA, USA).
Statistical analysis
Statistical calculations were examined by one-way analysis of variance (ANOVA), followed by Tukey's post hoc range test using SPSS Statistics 23 (SPSS, Inc., Chicago, IL, USA). The data are presented as mean ± standard error of duplicates to quadruplicates (n = 2–4). Significance is presented in each figure legend.
Results
Halleridone does not have significant cytotoxicity at <40 μM
Before examining how halleridone affected cellular signaling transduction, halleridone cytotoxicity was measured using the HT22 (Fig. 1B) and SH-SY5Y (Fig. 1C) cell lines. Halleridone did not show significant cytotoxicity at 10–40 μM in either HT22 or SH-SY5Y cell lines (Figs. 1B and 1C). Treatment with 10–40 μM halleridone in HT22 cells increased the phosphorylation of PI3K (Y199) and Akt (S473) in a concentration-dependent manner. In particular, 40 μM halleridone significantly activated the phosphorylation of PI3K and Akt (Supplementary Fig. S1; Supplementary Data are available online at
Halleridone changes Nrf2 expression and movement
The expression pattern of nuclear Nrf2 was evaluated by Western blot to investigate the action of Nrf2 by halleridone in the murine hippocampal HT22 cell line. Halleridone treatment at 40 μM significantly increased the protein expression of total Nrf2 and nuclear Nrf2 by up to 2.2- and 2.9-fold, respectively, compared with untreated cells (Figs. 2A, B). Moreover, halleridone treatment upregulated the expression of Nrf2 in the cytoplasm and nucleus and particularly induced Nrf2 translocation from the cytoplasm to the nucleus. However, Keap1 protein expression, as examined through immunocytochemistry, was slightly decreased by treatment with 40 μM halleridone (Fig. 2C).
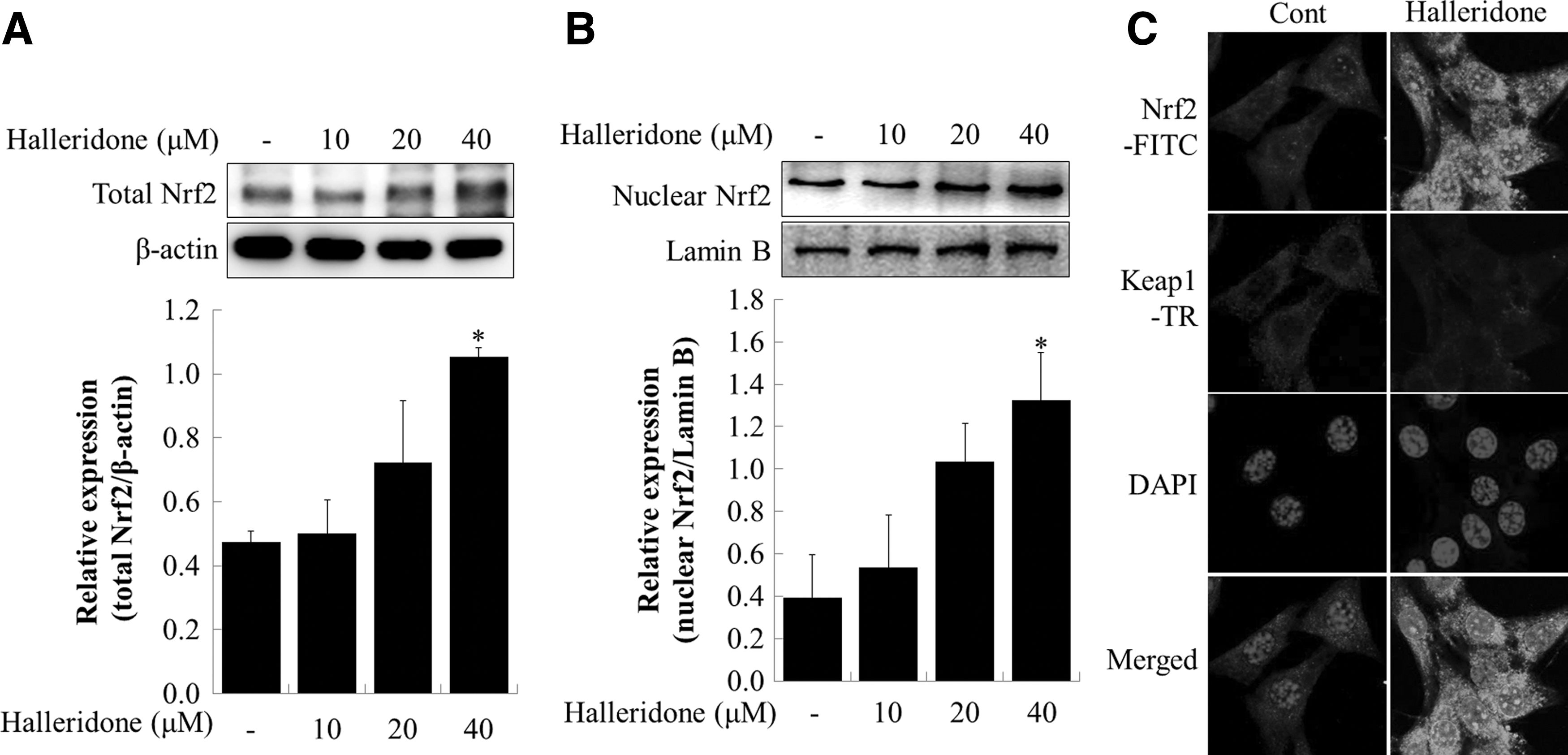
Effect of halleridone on the Nrf2 signaling pathway in HT22 cells.
Halleridone increases the transcriptional activity of the ARE gene in HT22-ARE cells
Brusatol, which is an Nrf2 inhibitor known to rapidly and transiently deplete Nrf2 protein, 19 was used to pretreat HT22 cells 2 h before halleridone treatment to examine the Nrf2-mediated ARE transcriptional activity by halleridone. Halleridone induced ARE transcriptional activity at 10, 20, and 40 μM in a concentration-dependent manner, whereas these activities were sharply curtailed to around half of those levels with 100 nM brusatol pretreatment (Fig. 3A). These results indicate that ARE gene transcriptional activation induced by halleridone treatment is mediated by Nrf2. As shown in Figure 3B, halleridone treatment upregulated mRNA expression levels by 15.9-fold compared with controls. Halleridone treatment significantly increased HO-1 expression levels by up to 2.8-fold at 40 μM. HO-1 expression was slightly decreased by 100 nM brusatol treatment (Fig. 3C).

The effect of halleridone on ARE gene transcriptional activity and HO-1 expression.
Halleridone reduces microglial Aβ 1-42 expression possibly through Nrf2 activation
The effects of halleridone on anti-inflammatory response and Aβ 1-42 clearance were elucidated by transiently transfecting plasmids containing Aβ 1-42 and the pEGFP gene to BV-2 cells. Then, the transfected BV-2 cells were treated with halleridone. Aβ 1-42 was overexpressed by transfection with pEGFP-C1/Aβ 1-42 plasmids in the BV-2 cells, and green fluorescence intensity was diminished by 40 μM halleridone treatment. Moreover, the pretreatment of brusatol before halleridone treatment suppressed the halleridone-induced decrease in green fluorescence intensity (Fig. 4).
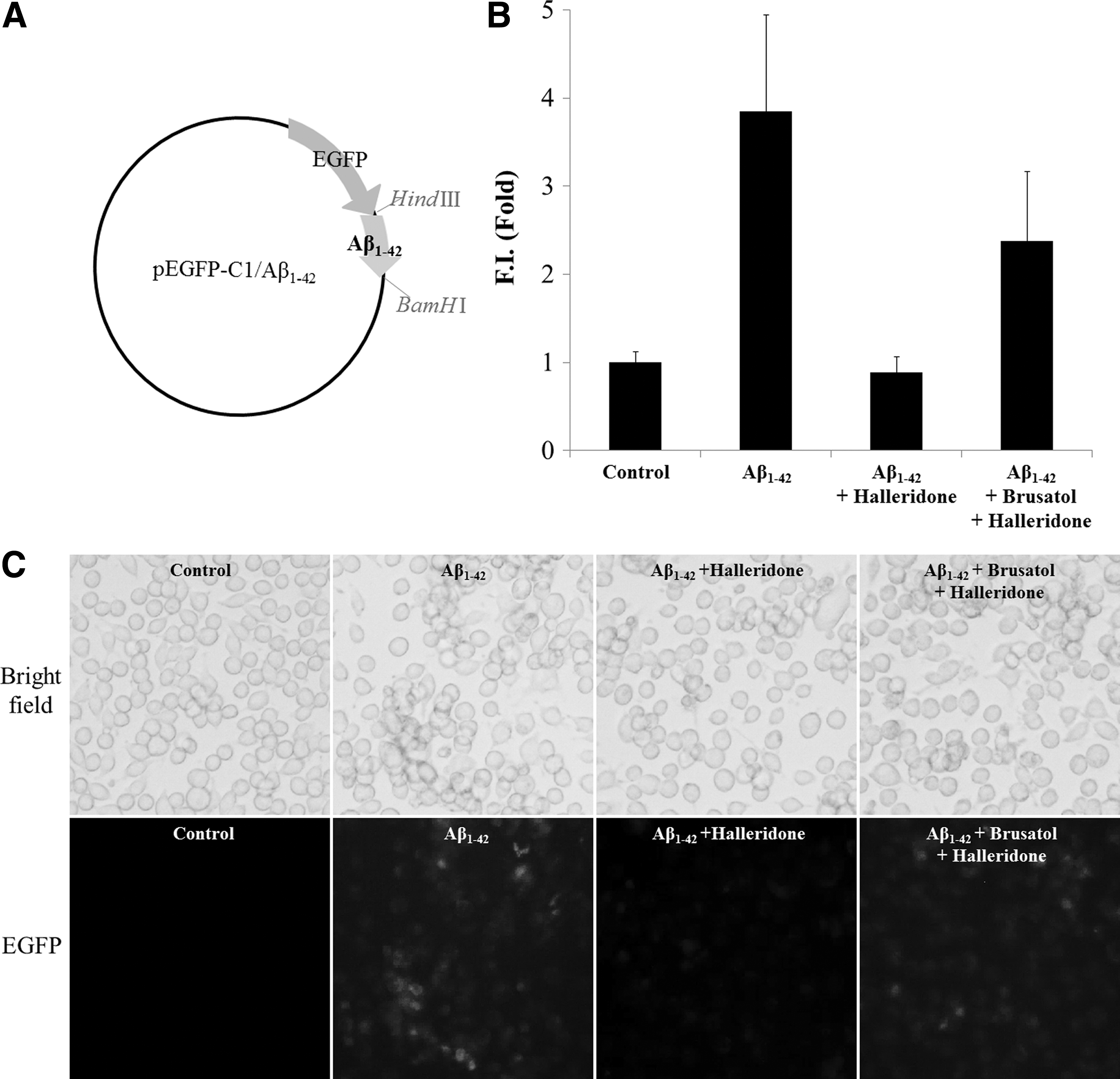
Effect of halleridone and/or brusatol on Aβ
1-42 peptides in BV-2 cells. BV-2 cells were transfected with 1.0 μg of Aβ
1-42 plasmids for 12 h. The medium was discarded and replaced with phenol red-free medium in the absence or presence of 100 nM brusatol. After 2 h, the medium was replaced with fresh medium containing 40 μM halleridone.
Halleridone downregulates nuclear pNF-κB-mediated inflammatory protein expression induced by Aβ 1-42 transfection in microglial BV-2 cells
As shown in Figure 5A, pNF-κB (S311) accumulated in the nuclei of BV-2 cells transfected with 2.0 μg of Aβ 1-42. This nuclear accumulation of pNF-κB was reduced by halleridone treatment at concentrations of 10–40 μM. The cellular expression of downstream inflammatory proteins, including inducible NO synthase (i-NOS) and cyclooxygenase II (COX-II), increased after Aβ 1-42 transfection. However, halleridone treatment significantly reduced the expression of i-NOS and COX-II (Fig. 5B).
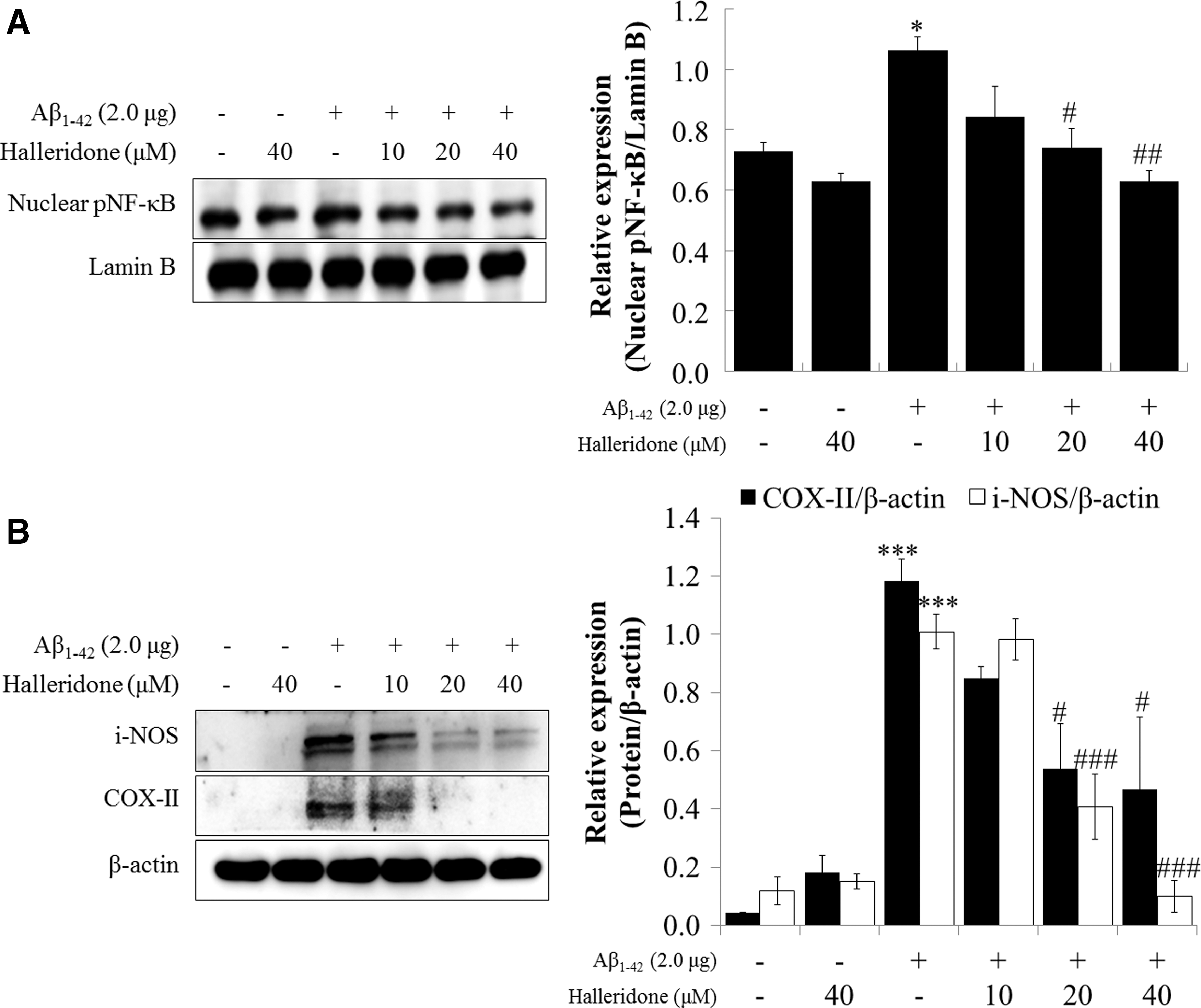
The effect of halleridone on the expression of pNF-κB or inflammatory proteins in BV-2 cells. BV-2 cells were seeded, transfected with Aβ
1-42 plasmids for 12 h, and the medium was than replaced with fresh phenol red-free medium containing halleridone for 24 h.
Halleridone decreases inflammatory cytokines and NO in the BV-2 cell line
Cytokine assays were performed to determine the levels of secreted IL-6 and IL-1β in BV-2-conditioned medium. As indicated in Figures 6A and B, Aβ 1-42 significantly increased the levels of IL-6 and IL-1β in media. When treated with halleridone, it effectively and dramatically reduced Aβ 1-42-induced IL-6 and IL-1β levels. For PGE2, which is a principal mediator of inflammation, halleridone significantly decreased PGE2 levels that were elevated by Aβ 1-42 transfection in the conditioned medium of BV-2 cells. NO, which is a biomarker in the final steps in inflammatory responses, was escalated by Aβ 1-42 transfection. Halleridone also lowered the secreted NO levels in conditioned medium in a concentration-dependent manner.
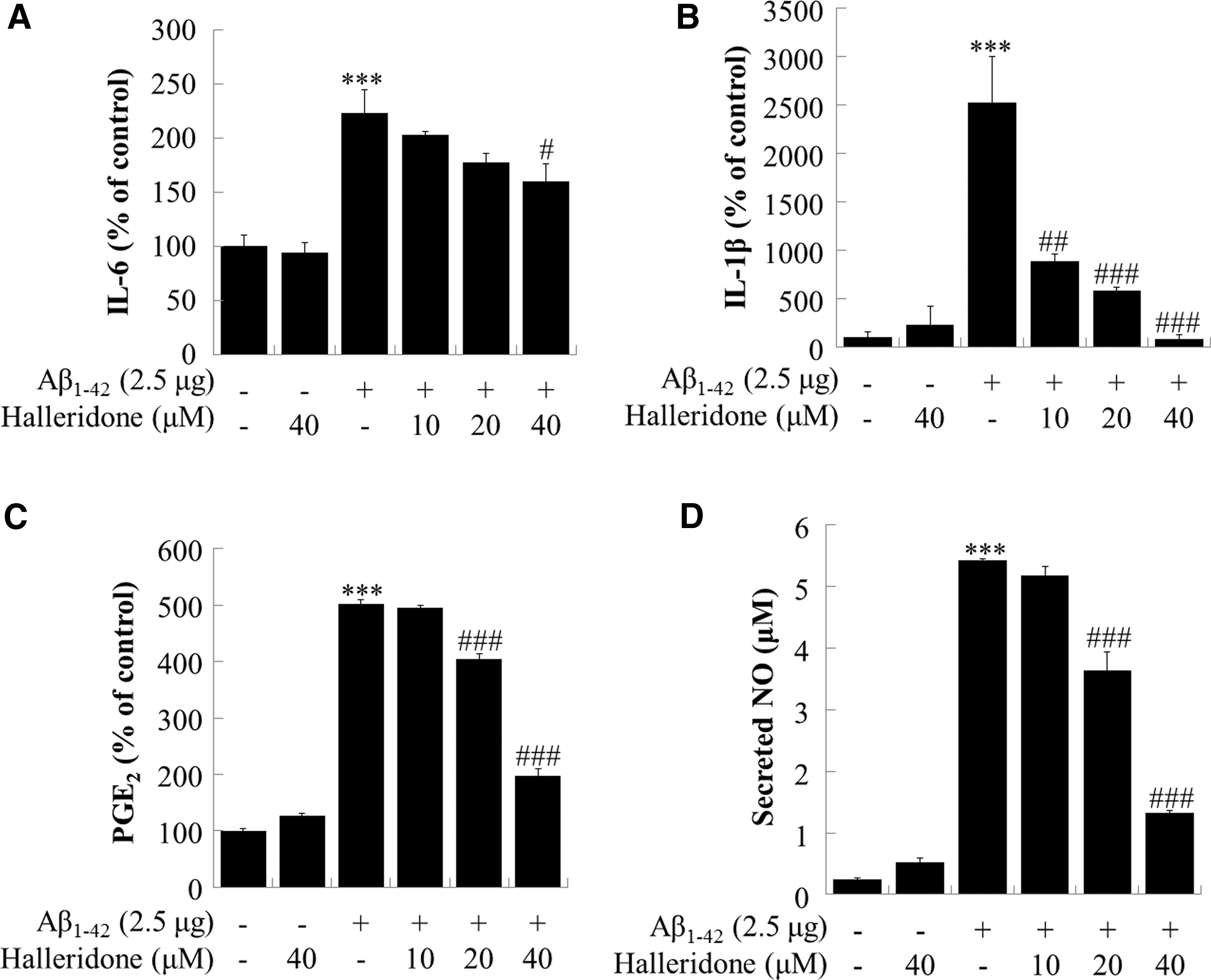
Effect of halleridone on inflammatory cytokines, PGE2, and NO secretion from BV-2 cells. BV-2 cells were seeded, transfected with Aβ
1-42 plasmids for 12 h, and the medium was then replaced with fresh phenol red-free medium containing various concentrations of halleridone for 24 h. The conditioned medium was collected and subjected to analysis for
Discussion
The PI3K/Akt signaling pathway is essential for both neuroprotection and cell survival and acts as a regulator of antiapoptotic proteins such as those of the Bcl-2 family. 20 –23 Additionally, the PI3K/Akt signaling pathway mediates the nuclear translocation of Nrf2 by phosphorylation in several cell types, including neuronal cells. 9,24 Under normal conditions, Nrf2 is mostly present in the cytoplasm of hippocampal neurons and not much in the nucleus. 25 Hence, Nrf2 is released from Keap1 due to mutation, DNA methylation, decreased Keap1 expression, and Keap1 succination under stressed condition. The detachment of Nrf2 from Keap1 inhibits Nrf2 ubiquitination and the action of disruptor proteins. 26 Many studies have reported that released Nrf2 sequentially binds to the promoter region of ARE sequence, activates target genes, and translates antioxidant enzymes, including HO-1. 27
HO-1, one of the marker proteins of antioxidant enzymes, is an inducible enzyme that responds to oxidative stress. Heme is degraded into the strong antioxidants, biliverdin and bilirubin, by HO-1. Therefore, HO-1 is considered a therapeutic target in neurodegenerative diseases such as AD. 28
Our results demonstrate that halleridone significantly influenced pPI3K, pAKT, total Nrf2, nuclear Nrf2, and HO-1 expression at 40 μM concentration compared with the vehicle groups. Moreover, in silico results revealed that halleridone interacted with the bric-a-brac/tramtrack/broad (BTB) domain of Keap1 mostly through van der Waals interaction and conventional hydrogen bonding to Gly148 in Keap1 (Supplementary Fig. S6). Among the 25 cysteines in murine Keap1 and 27 cysteines in human Keap1, Cys151 presented in the BTB domain and Cys273 and Cys288 react with oxidants or electrophiles. These reactions result in Nrf2 release from Keap1. 29 –31 These results suggest that the increase of Nrf2 in the nucleus by halleridone might be associated with pPI3K/AKT and Keap1 inhibition through Cys151. However, the specific underlying mechanism remains unknown.
Neuroinflammation studies on Aβ clearance in microglia have received much attention. The phagocytic activity of microglia appears to be involved with Aβ clearance. 32 The pNF-κB signaling pathway is promoted by pathogenic molecules such as Aβ and can lead to microglial activation by inducing inflammation. The elevated secretion of proinflammatory cytokines such as IL-6 and IL-1β is observed in AD patients. 33 Halleridone reduced Aβ 1-42 overexpression, as indicated by the green fluorescence in Figure 4B. However, the green fluorescence was reversed by brusatol pretreatment. These results indicate that halleridone could reduce AD pathology at the cellular levels and that Nrf2 is partially associated with the effect of halleridone on Aβ 1-42 reduction.
Autophagy, 34 caspase 3 cascades, 35 and proteasomal degradation 36 could be possible Aβ 1-42 clearing mechanisms. However, technical limitations have prevented clear elucidation of the mechanism by which Aβ 1-42 reduction is mediated by intracellular clearance of halleridone. Nrf2/Keap1 signaling can affect Nrf2-mediated inflammation; however, this finding is controversial. Additionally, several recent articles have reported that Keap1 regulates inflammatory signaling. Our in silico results have demonstrated that halleridone can strongly interact with the BTB domain of Keap1, which inhibits Nrf2. Even though these data do not illustrate the overall Nrf2/Keap1 signaling mechanism through experimental results, the data allow us to determine the involvement of halleridone in Nrf2/Keap1-related anti-inflammation.
In summary, halleridone activated the pPI3K/pAkt signaling pathway and upregulated Nrf2/ARE-mediated HO-1 expression in murine hippocampal HT22 cells. Halleridone promoted effective intracellular Aβ 1-42 peptide reduction and this activity may be partially due to Nrf2 action. Halleridone also downregulated pNF-κB signals and inflammatory responses elevated by Aβ 1-42 overexpression. In conclusion, halleridone upregulated Nrf2-mediated HO-1 expression in HT22 cells and exerted antineuroinflammatory activity through Nrf2/Keap1 and pNF-κB signaling in microglia BV-2 cells (Supplementary Fig. S10). These results indicate that halleridone may be utilized in AD research with regard to mitigating oxidative stress and neuroinflammation.
Footnotes
Acknowledgments
This work was supported, in part, by grants from the Marine Biotechnology Program of the Ministry of Oceans and Fisheries (PJT200669) and from the Bio and Technology Development (2016 M3A9A5919548) of the National Research Foundation of Korea (NRF), which is funded by the Ministry of Science, Information and Communication Technology (ICT) and Future Planning.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.