
Abstract
Red ginseng oil (RGO) has been shown to possess anti-inflammatory and hepatoprotective activity. In this study, we evaluated the inhibitory effect of RGO on 12-O-tetradecanoylphorbol-13-acetate (TPA)-stimulated neoplastic transformation of JB6 P+ cells. RGO pretreatment abolished the transformation of JB6 P+ cells challenged by TPA. RGO suppressed the transactivation of activator protein-1 (AP-1) and nuclear factor kappa B (NF-κB) transcription factors as well as protein levels of cyclooxygenase-2, cyclin D1, cyclin E, and Bcl-2 in the TPA-treated cells. Additionally, TPA-induced phosphorylations of extracellular signal-regulated kinases, 90 kDa ribosomal S6 kinase 2, c-Jun N-terminal kinases, and glycogen synthase kinase 3β were downregulated in the presence of RGO. Furthermore, RGO induced the nuclear factor erythroid 2-related factor 2 (Nrf2)-mediated antioxidant enzyme heme oxygenase-1 (HO-1) expression, and effectively blocked the overproduction of TPA-induced reactive oxygen species. These results suggest that RGO exerts a potent chemopreventive activity in skin cell model.
Introduction
C
Along with numerous biological properties of water-soluble fractions of red ginseng, studies on its lipid-soluble fraction have recently identified several pharmacological activities. A previous report has demonstrated that a lipid-soluble red ginseng extract exhibits and blocks human lung tumor growth in xenograft models. 4 Lee et al. have documented that the lipid-soluble fraction of ginseng marc extracted by a sequential extraction process has anticancer activity. 5 Our studies have also evaluated the pharmacological properties and the molecular mechanisms of red ginseng oil (RGO), which is extracted using a supercritical CO2 fluid extraction. RGO exhibited cytoprotective effects in H2O2-treated HepG2 cells as well as carbon tetrachloride (CCl4)-treated mice through the upregulation of antioxidant defense systems. 6 RGO also showed anti-inflammatory activity through blocking secretion of proinflammatory mediators and cytokines in lipopolysaccharide-induced RAW 264.7 cells. 7
RGO contained linoleic acid, β-sitosterol, and bicyclo-(10.1.0)tridec-1-ene as its major components and displayed chemopreventive properties by the induction of nuclear factor erythroid 2-related factor 2 (Nrf2)/antioxidant response element (ARE)-mediated cytoprotective genes in HepG2 cells. 8 Furthermore, RGO was shown to be safe and nontoxic at 5000 mg/kg body weight in an acute toxicity rat model. 9 Although a few reports have shown anticancer properties of hexane-extracted lipid-soluble fractions from red ginseng, there has been no information on chemopreventive effect of supercritical CO2 fluid-extracted RGO in a skin carcinogenesis model.
JB6 P+ epidermal cell line is a useful model for investigating molecular mechanisms and signaling pathways linked to tumor promotion of skin carcinogenesis as well as elucidating chemopreventive properties of therapeutic compounds. 10 Exposure of JB6 P+ cells to chemical irritants, such as 12-O-tetradecanoylphorbol-13-acetate (TPA), epidermal growth factors, or ultraviolet B (UVB), leads to neoplastic transformation. 11,12 Therefore, this study was conducted to examine the effect of RGO on the TPA-induced neoplastic transformation of JB6 P+ mouse epidermal cells and identify its molecular mechanisms.
Materials and Methods
Chemicals
Paraformaldehyde, Triton X-100, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), and 4′,6′-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against cyclin E, Nrf2, β-actin, and horseradish peroxidase-conjugated anti-goat were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cyclin D1, p-ERK, p-JNK, p-GSK3β, p-p90RSK, and horseradish peroxidase-conjugated anti-rabbit antibodies were supplied by Cell Signaling Technology (Beverly, MA, USA). All other reagents were of the highest reagent grade. RGO was prepared as described in the previous study. 8 RGO was dissolved by complexing with bovine serum albumin (BSA). RGO dissolved in ethanol was very slowly added dropwise to the 5% BSA solution. Vehicle was composed of 5% BSA with an equivalent volume of ethanol in RGO stocks. The final ethanol concentration in all treatments was less than 0.2% v/v.
Cell culture
JB6 Cl41-5a (JB6 P+) mouse epidermal cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The JB6 P+ cells were maintained in Eagle's minimal essential medium (MEM) supplying 5% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin and incubated in a 5% CO2 incubator at 37°C.
Cell viability
JB6 P+ cells (104 cells/well) were cultured in 96-well plates for 24 h to allow growth. The cells were treated with an array of RGO concentrations (0–200 μg/mL) for 24 h. The cell viability was measured using MTT assay. The quantity of formazan in each well was measured by recording absorbance at 570 nm with a microplate reader (BioTek Instruments, Winooski, VT, USA).
Measurement of intracellular production of reactive oxygen species
The intracellular reactive oxygen species (ROS) accumulation was measured using a fluorescent probe dichloro-dihydro-fluorescein diacetate (DCFH-DA). Briefly, after starvation, JB6 P+ cells were pretreated with RGO for 24 h and then treated with TPA (20 ng/mL) for additional 12 h. The cells were washed twice with phosphate-buffered saline (PBS) and incubated with 20 μM DCFH-DA for 1 h at 37°C. Fluorescence intensity was recorded using a fluorescence microplate reader (Synergy HT; BioTek Instruments) at 485/20 nm excitation and 528/20 nm emission.
Anchorage-independent transformation assay
JB6 P+ cell transformation was evaluated using an anchorage-independent transformation assay. Briefly, the cells (8 × 103 cells/well) were exposed to TPA (20 ng/mL) with/without RGO (0–50 μg/mL) in the 0.33% basal Eagle's medium agar (top layer) containing 10% FBS over 0.5% basal Eagle's medium (bottom layer) supplying 10% FBS. Soft agar cultures were incubated in a 5% CO2 incubator at 37°C for 14 days and cell clones were photographed using a digital camera (Paxcam, IA, USA).
Confocal microscopy analysis
JB6 P+ cells were seeded in the coverglass-bottom dishes. After starvation with medium containing 0.1% FBS for 24 h, the cells were pretreated with 50 μg/mL RGO and then treated with 20 ng/mL TPA for 1 h. The cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, and subsequently blocked with 3% BSA in PBS for 2 h. The cells were incubated with anti-p65 and anti-c-Jun antibodies for 2 h and then hybridized with an Alexa Fluor 555-conjugated secondary antibody for additional 2 h in the darkness. After washing twice with PBS and incubating with 1 μg/mL of DAPI for 20 min in the darkness, images were visualized using a LSM 510 laser confocal microscope (Zeiss, Jena, Germany).
Western blot analysis
Equal amounts of total protein measured by the BCA Protein Assay Kit (Pierce, Rockford, IL, USA) were separated on SDS-PAGE gels. The protein was then transferred onto polyvinylidene fluoride membranes using a semidry transfer system (Bio-Rad, Hercules, CA, USA). After blocking with 5% nonfat milk, the membranes were hybridized with appropriate specific primary antibodies at 4°C for overnight and subsequently incubated with anti-rabbit or anti-goat secondary antibodies for 3 h at 4°C. Protein bands were visualized onto X-ray film using western blotting Luminol reagent (Santa Cruz Biotechnology).
Statistical analysis
Results are displayed as mean ± standard deviation. Student's t-test was used for analysis of significance between control and treated groups. A value of P < .05 was considered as statistically significant.
Results
Effect of RGO on the viability of JB6 P+ mouse epidermal cells
To evaluate the effect of RGO on the viability of JB6 P+ cells, the cells were incubated with a range of RGO concentrations (0–200 μg/mL) and cell survival was determined using MTT assay. As shown in Figure 1, treatment of RGO resulted in a slight decrease of JB6 P+ cell survival in a dose-dependent fashion. The highest concentration of RGO (200 μg/mL) reduced cell viability by ∼25%. Treatment of RGO up to 50 μg/mL that did not produce a significant toxic effect on JB6 P+ cells was used for further experiments.
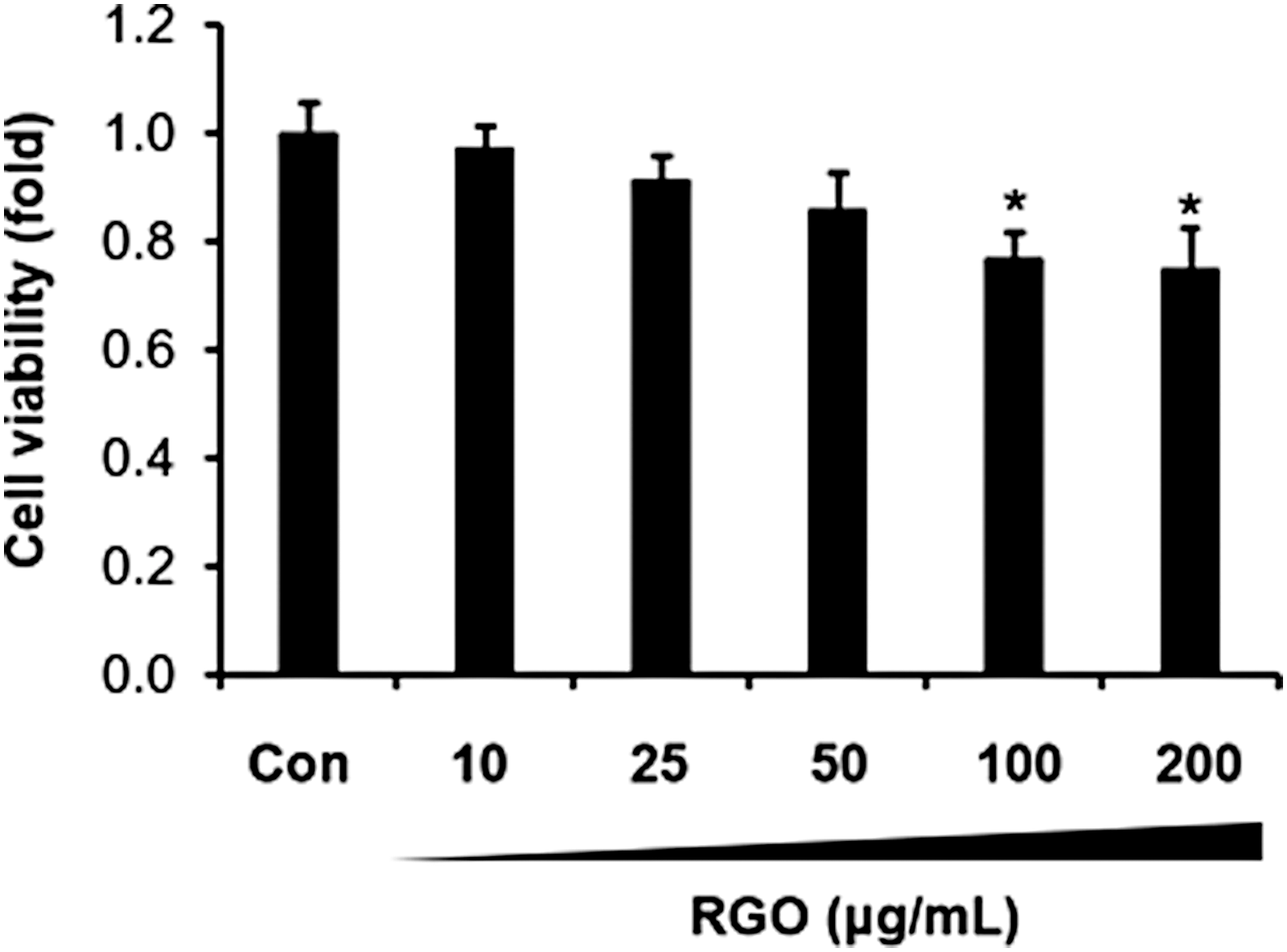
Effect of RGO on the JB6 P+ cell viability. JB6 P+ cells (1 × 104) were seeded in 96-well plates in MEM containing 5% FBS, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a 5% CO2 incubator for 24 h. The cells were treated with various concentrations of RGO (0–200 μg/mL) for 24 h and followed by adding MTT solution for 4 h at 37°C. Absorbance was recorded at 570 nm. Data are presented as the mean ± SD of three independent experiments. *P < .05 is significantly different as compared with the control. FBS, fetal bovine serum; MEM, Eagle's minimal essential medium; MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; RGO, red ginseng oil; SD, standard deviation.
RGO suppresses TPA-induced neoplastic cell transformation
JB6 P+ mouse epidermal cell line is a unique model to study the molecular mechanisms of tumor promotion and investigate antitumor-promoting agents. Thus, JB6 P+ cells were employed to evaluate the effect of RGO on TPA-induced neoplastic cell transformation. RGO treatment significantly suppressed TPA-stimulated JB6 P+ cell transformation in a dose-dependent fashion (Fig. 2). RGO at 50 μg/mL reduced the number of colonies by 80% when compared with the TPA-treated cells.
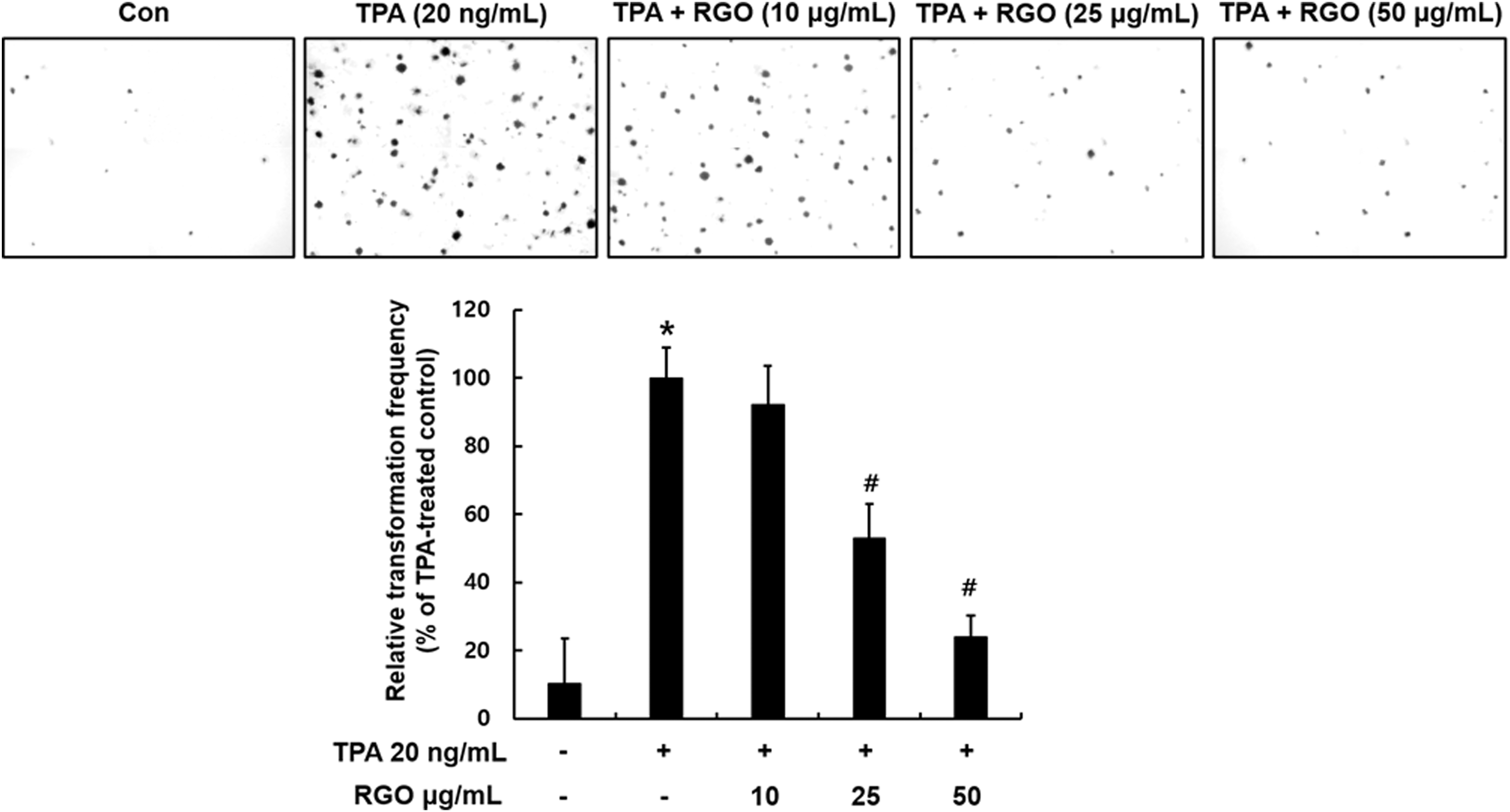
Effect of RGO on TPA-induced neoplastic transformation of JB6 P+ cells. Cells (8 × 103) treated with/without RGO (10, 25, and 50 μg/mL) were exposed to TPA (20 ng/mL) in 0.33% basal medium Eagle's agar containing 10% FBS. The plates were maintained at 37°C in a 5% CO2 incubator for 14 days. The colonies were photographed using a digital camera at 40 × magnification. Data are presented as the mean ± SD of three independent experiments. *P < .05, is significantly different as compared with the control. # P < .05 is significantly different as compared with the TPA-induced cells. TPA, 12-O-tetradecanoylphorbol-13-acetate.
RGO inhibits activator protein-1 and nuclear factor kappa B transactivation in TPA-stimulated JB6 P+ cells
Next, we determined the effect of RGO on the activation of activator protein-1 (AP-1) and nuclear factor kappa B (NF-κB) pathways in the TPA-exposed JB6 P+ cells. Pretreatment with RGO significantly inhibited the phosphorylation of c-Jun strongly in cells enhanced by TPA challenge (Fig. 3A). In addition, TPA-induced nuclear translocation of c-Jun was reduced by the presence of RGO (50 μg/mL) (Fig. 3B). Along with the activation of c-Jun/AP-1 pathway, p65/NF-κB pathway was also activated by TPA exposure through the enhanced phosphorylation of p65, which was significantly decreased by RGO preincubation (Fig. 3C). Consequently, confocal analysis indicated that TPA-enhanced p65 nuclear translocation decreased by pretreatment of RGO (50 μg/mL) (Fig. 3D). These results suggest that RGO inhibits JB6 P+ cell neoplastic transformation possibly through blocking AP-1 and NF-κB pathways.
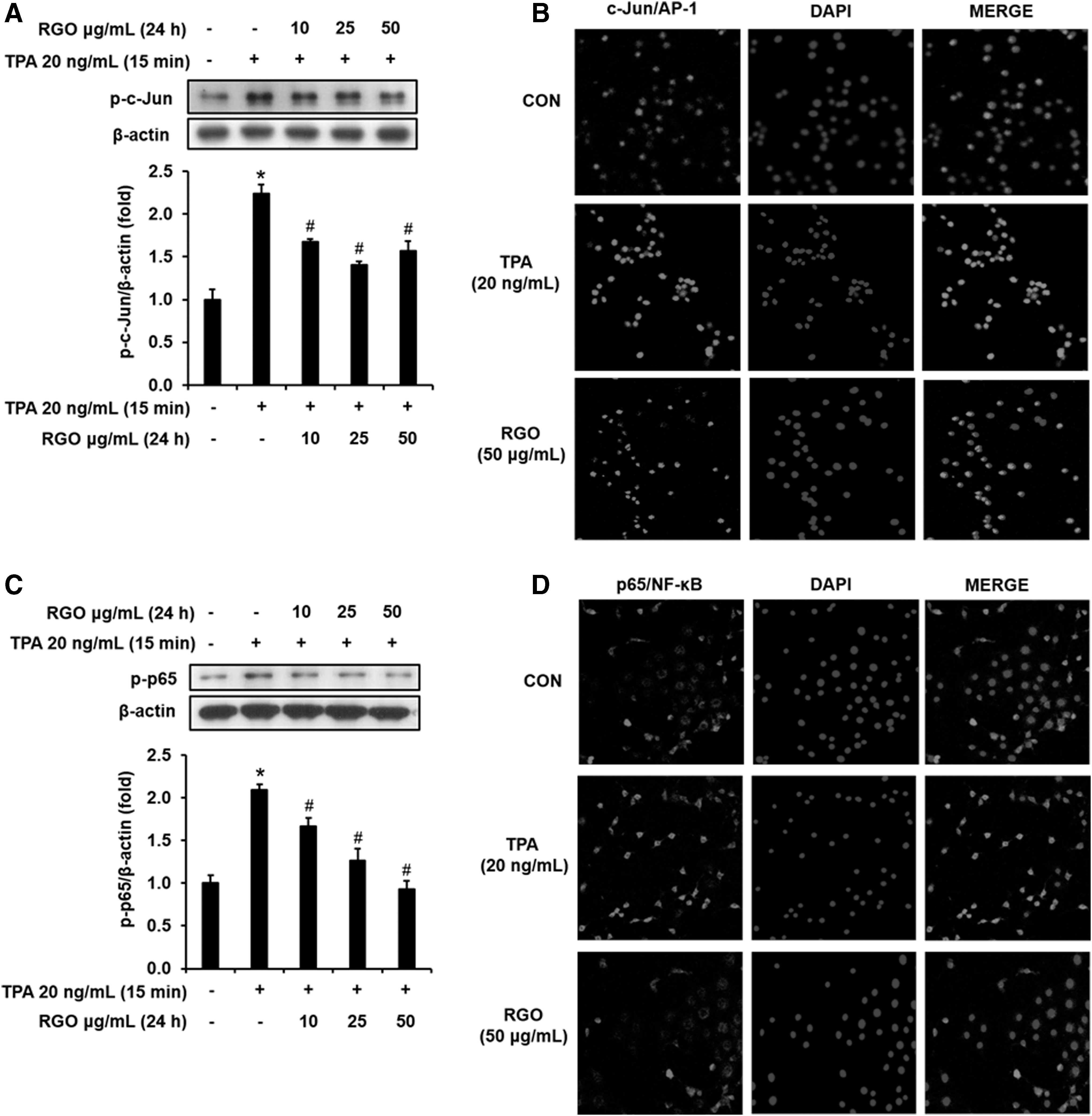
Effect of RGO on the activation of c-Jun/AP-1 and p65/NF-κB in the TPA-treated JB6 P+ cells.
RGO attenuates protein expression of COX-2, cyclin D1, cyclin E, and Bcl-2 in TPA-treated JB6 P+ cells
We also analyzed the effects of RGO on a series of molecular targets associated with inflammation, cell cycle, proliferation, and angiogenesis. COX-2 expression was upregulated about twofold by TPA (20 ng/mL) exposure when compared with normal control. However, pretreatment with RGO remarkably and dose dependently downregulated the TPA-stimulated expression level of COX-2 in JB6 P+ cells (Fig. 4A). In accordance with the anchorage-independent cell growth assay, RGO pretreatment significantly reduced the TPA-induced protein expressions of cyclin D1 and cyclin E, which are associated with cell cycle and cell proliferation. Especially, RGO at 50 μg/mL exhibited the greatest inhibitory effect on the cyclin D1 and cyclin E expressions, suggesting promotion of G0/G1 phase arrest and inhibition of neoplastic transformation of JB6 P+ cells (Fig. 4B). Elevated level of antiapoptotic protein Bcl-2 was observed in response to TPA treatment, whereas preincubation of RGO suppressed Bcl-2 expression in the TPA-treated JB6 P+ cells (Fig. 4C).
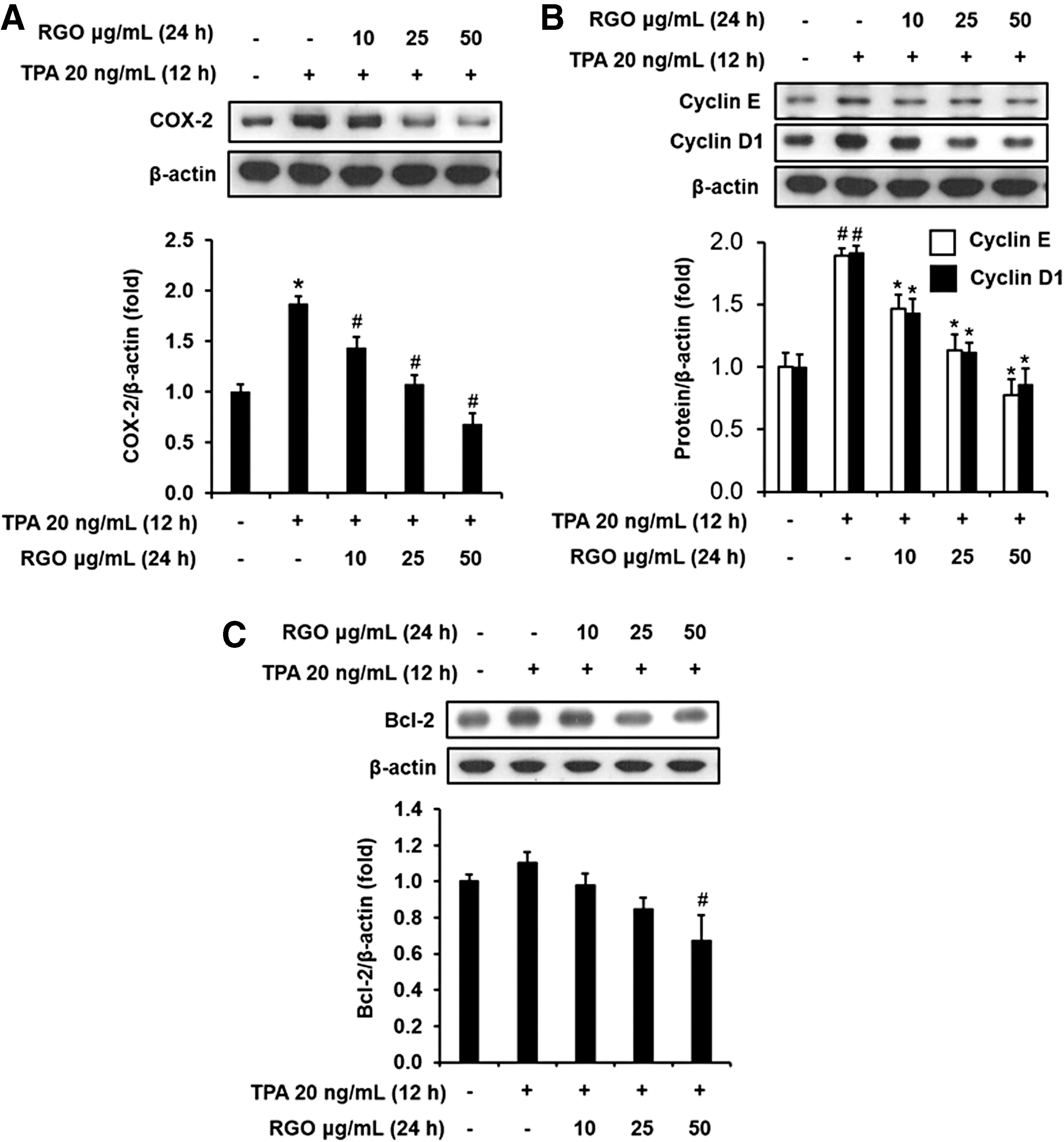
Effect of RGO on the protein expression of
RGO inhibits upstream kinases in TPA-treated JB6 P+ cells
Next, we further elucidated the effect of RGO on the phosphorylation of upstream kinases, such as ERK, p90RSK, JNK, and GSK-3β, which are well known to contribute to cell transformation, and AP-1 and NF-κB transaction in JB6 P+ cells. As illustrated in Figure 5A, TPA treatment activated the phosphorylations of ERK/p90RSK2. However, TPA-enhanced phosphorylations of ERK and p90RSK2 were attenuated by pretreatment with RGO. TPA exposure also dramatically enhanced phosphorylated JNK level, which was suppressed by the presence of RGO in the cells (Fig. 5B). Along with the inhibition of ERK/p90RSK2 and JNK signaling pathways, TPA-stimulated phosphorylation of GSK-3β was abolished dose dependently by preincubation of RGO (Fig. 5C).
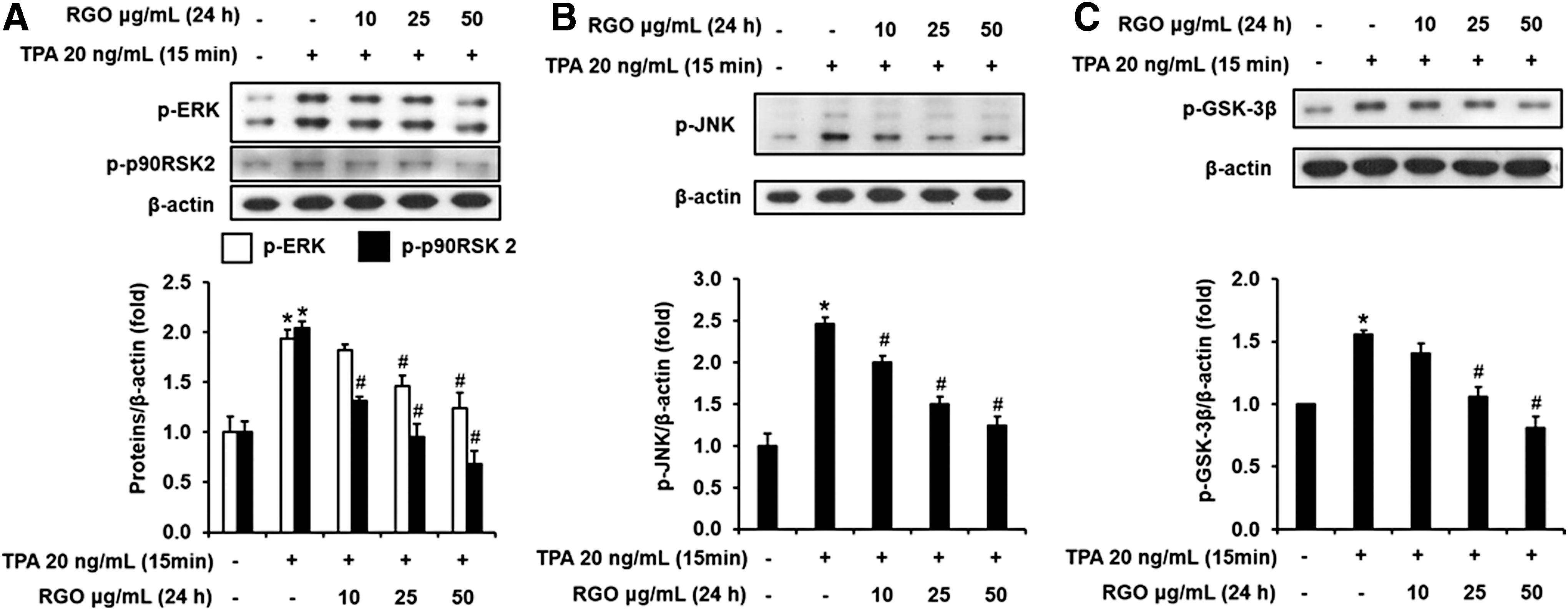
Effect of RGO on the phosphorylation levels of
RGO induces Nrf2-mediated heme oxygenase-1 expression and protects against TPA-induced ROS accumulation
Transcription factor Nrf2 takes part in regulating various phase II detoxifying and antioxidant enzymes, including heme oxygenase-1(HO-1). When the JB6 P+ cells were treated with RGO, Nrf2 protein expression was time dependently upregulated by RGO treatment (Fig. 6A). The expression of Nrf2 started to significantly increase after 2 h and was continuously elevated until 24 h treatment of RGO at 50 μg/mL. In addition to Nrf2 expression, RGO also elevated HO-1 expression, an antioxidant enzyme (Fig. 6B). To confirm whether this activation of Nrf2/HO-1 antioxidant machinery by RGO can provide antioxidant capacity, we measured the TPA-stimulated ROS formation in the JB6 P+ cells with and without RGO. Pretreatment of cells with RGO significantly attenuated the ROS overproduction caused by TPA (Fig. 6C), suggesting that the increased level of HO-1 by RGO might contribute to the elimination of ROS as well as the suppression of JB6 P+ cells transformation.
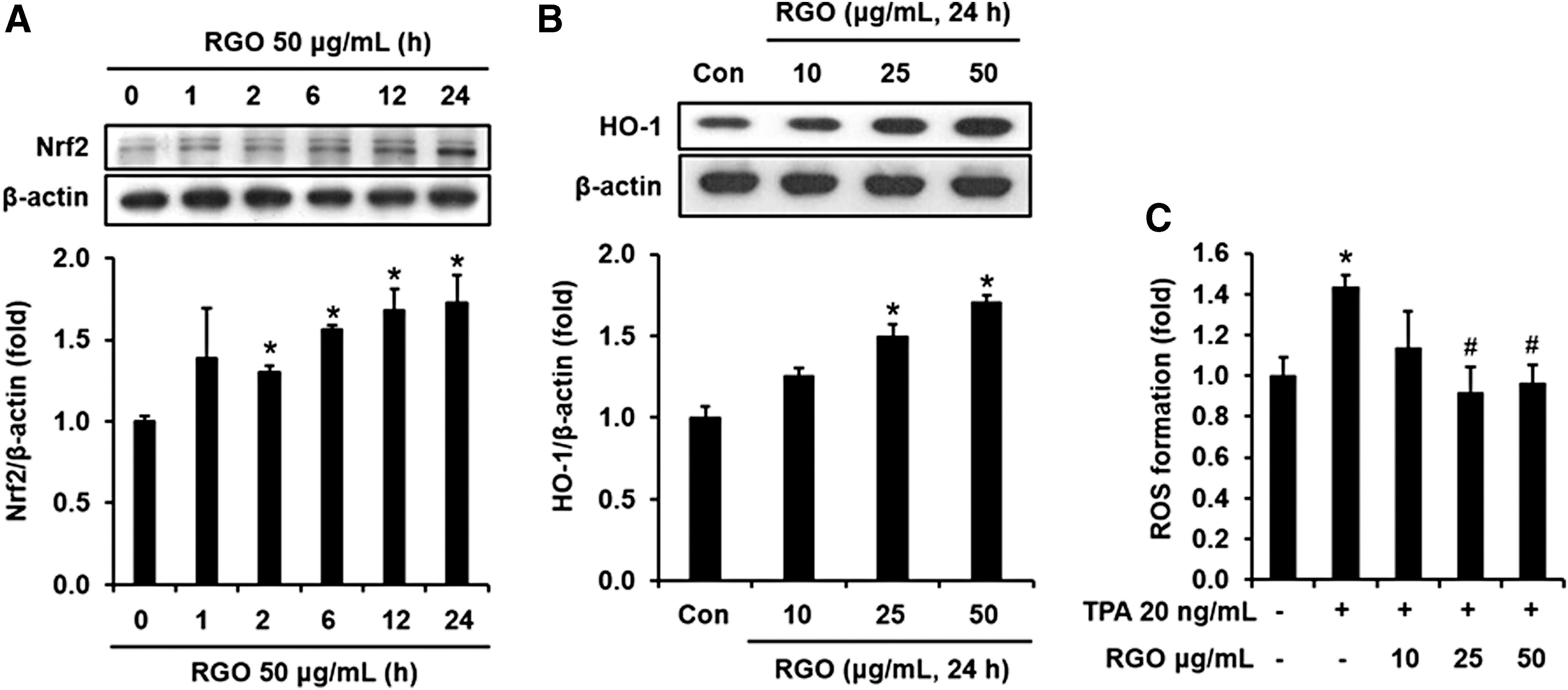
Antioxidant activity of RGO in the JB6 P+ cells.
Discussion
Recently, naturally occurring phytochemicals are attracting great interest owing to their potential health benefits in preventing or suppressing the initiation and subsequent progression of cancer. The present study revealed that RGO might be a potent chemopreventive agent against skin cancer cell transformation and tumorigenesis. Our previous study has indicated that major components of RGO are linoleic acid, sitosterol, and bicyclo(10.1.0)tridec-1-ene, constituting more than 70% of RGO by GC/MS analysis. 8 Although chemopreventive properties of bicyclo(10.1.0)tridec-1-ene have not been investigated, β-sitosterol has been demonstrated to have antitumor activity. 13 Recent studies have shown that β-sitosterol promotes cell cycle arrest and apoptosis in breast cancer cells, 14 and prostate cancer cells. 15 β-sitosterol exerts inhibitory effect on the proliferation of human gastric adenocarcinoma cells and tumor growth in tumor xenograft. 16 Additionally, β-sitosterol has been reported to suppress neoplastic transformation of JB6 P+ cells exposed to TPA. 17 Topical treatment of β-sitosterol also decreases the TPA-induced inflammation in mouse models. 18,19 While linoleic acid is unlikely to block TPA-stimulated JB6 P+ cell transformation as well as skin tumor promotion in mice, a lipoxygenase metabolite of linoleic acid suppresses TPA-induced inflammation as well as tumor promotion. 20 Therefore, the chemopreventive property of RGO in the current skin model might be at least associated with the presence of β-sitosterol as a major component.
Transcription factors, including AP-1 and NF-κB, play a vital role in the various pathological conditions such as inflammation, neoplastic transformation, tumorigenesis, angiogenesis, metastasis, and invasion. 21,22 Many studies have reported that both AP-1 and NF-κB may be implicated in the TPA-stimulated JB6 P+ cell transformation. 23,24 The AP-1 transcription factor is a protein dimer composed of Jun (c-Jun, JunB, and JunD) and Fos (c-Fos, Fra-1, and Fra-2) families, and transcriptionally drives target genes involving in tumorigenesis and preservation of the transformed state through collaborating with the TPA-responsive element. 25,26 Suppression of AP-1 transactivation has been demonstrated to inhibit TPA-induced transformation of cancer cells. 27,28 Moreover, it has been shown that transgenic mice expressing transactivation mutant c-jun (TAM67) are protected from DMBA-TPA-caused skin papilloma genesis, suggesting that AP-1 is essential for tumorigenesis and a great target for prevention of tumor promotion as well as tumor progression. 29,30 Along with AP-1, NF-κB transcription factor is also required for transformation and tumor promotion of many different cancer cells. 31 Increased NF-κB activity is observed in the TPA-induced JB6 P+ cells transformation. 24 More interestingly, interaction of members of AP-1 (c-Jun or c-Fos) with component of NF-κB (p65) produces synergistic effects on both AP-1 and NF-κB-dependent gene activation. 32,33 Blockade of both AP-1 and NF-κB by various chemopreventive compounds has been found to block neoplastic transformation and tumor promotion. 34 In the present study, we showed that pretreatment with RGO decreased the phosphorylation and nuclear accumulation of c-Jun and p65, thereby preventing TPA-induced neoplastic transformation of JB6 P+ cells. Consequently, RGO pretreatment resulted in the downregulation of TPA-induced AP-1 and NF-κB-mediated target genes, including COX-2, Bcl-2, and cyclin D1 involving in the regulation of inflammation, cell transformation, cell growth, cell proliferation, and metastasis. COX-2 is considered as a mediator of inflammation and cancer through triggering inflammatory response and inducing neoplastic transformation as well as tumor progression. 35,36 Increased level of prostaglandins and/or overexpression of COX-2, which are found in many kinds of transformed cells, lead to blocking apoptotic cell death and potentiating the invasiveness of malignant cells. 37,38 Transgenic mice overexpressing the human cox-2 gene expressed a great incidence of focal mammary gland hyperplasia, dysplasia, and transformation into metastasis tumors. 39 Similarly, transgenic mice overexpressing cox-2 gene in their skin tissue resulted in epidermal hyperplasia and dysplasia. 40 In contrast, deficiency of cox-2 decreased skin tumorigenesis as well as intestinal tumors. 41 –43 Recently, many documents have shown that COX-2 enhances the TPA-induced JB6 P+ cell transformation and inhibition of COX-2 by variety of diverse agents prevents the neoplastic transformation of JB6 P+ cell and skin tumorigenesis. 36,44 In addition, Bcl-2 has been demonstrated to involve TPA-stimulated malignant transformation of mouse epithelial cells through enhancing AP-1 expression. 45 Cyclin D1 and cyclin E play a key role in regulating the growth phase of the cell cycle. Cyclin D1 is expressed in the middle of G1, whereas cyclin E expression is maximal at the G1-S transition. 46 Abnormal expression of cyclin D1 and cyclin E has been found in the development of many cancers, especially skin carcinogenesis. 47 –49 The cyclin D1-overexpressing mice exhibited increased sensitivity to a carcinogen. 50 Suppression of cyclin D1 has been indicated to attenuate the arsenite-induced transformation of human HaCaT cells. 51 Furthermore, cyclin D1-deficient mice have a decreased development of skin tumor. 52 Our study shows that along with the suppression of AP-1 and NF-κB transactivation, suppression of genes involving in the cell survival, cell proliferation, inflammation, and angiogenesis, such as Bcl-2, cyclin D1, cyclin E, and COX-2 contribute to the blocking effect of RGO on TPA-stimulated neoplastic transformation of JB6 P+ cells.
Accumulating evidences have indicated that the ERK pathway is linked to TPA-induced JB6 P+ cell transformation. 53,54 Abolishment of ERK activity by a specific inhibitor suppressed TPA-stimulated AP-1 and NF-κB transactivation and cell transformation. 55 In addition, p90RSK, a family of serine/threonine kinases regulated by ERK1/2, is also known to play an important function in transmitting signals for TPA-induced cell transformation and tumor promotion. 56 Tumor promoters such as TPA activate ERK1/2 and p90RSK phosphorylation, leading to stimulation of G1/S cell cycle transition and subsequent cell proliferation. 57,58 The activated ERKs/p90RSK axis can phosphorylate and activate AP-1 and NF-κB, thereby upregulating gene expression relating to cell transformation and tumor promotion. Also, previous documents have indicated that JNK activation also contributes to TPA-induced JB6 P+ cell transformation, and JNK inhibition by specific inhibitor is critical for blocking cell transformation. 43,58 In this study, RGO attenuated the phosphorylated levels of ERK, and p90RSK, and JNK the TPA-treated JB6 P+ cells. Additionally, GSK3β is a serine/threonine kinase that participates in the neoplastic transformation as well as tumor promotion. GSK3β is a negative regulator, where GSK3β activation (nonphosphorylated GSK3β at Ser 9 ) contributes to cell cycle arrest and apoptotic cell death, whereas GSK3β inactivation (phosphorylated GSK3β at Ser 9 ) results in the liberation of transcription factors relating to cell cycle/survival, proliferation, transformation, and tumor promotion. 59,60 Several studies have demonstrated a role of GSK3β as a “tumor suppressor” due to its negative regulation of AP-1 and NF-κB transactivation. 61,62 TPA has been demonstrated to induce neoplastic transformation of JB6 P+ cells through the phosphorylation of GSK3β. Therefore, abolishing GSK3β phosphorylation makes an important contribution to antitumor activity of RGO.
During the initiation of tumorigenesis, stresses caused by environmental toxins, mutagens, and carcinogens may directly or indirectly attack intracellular macromolecules, thereby resulting in mutation and dysfunction. 1 Elimination of ROS overproduction as well as toxicants before they damage crucial macromolecules is essential for protection of cells/tissues and for prevention of tumorigenesis. Nrf2 is regarded as a key regulator of a battery of these cytoprotective genes through its binding to ARE located in the promotor region of the genes. 63,64 Nrf2-knockout mice exhibited lower basal level of detoxification and antioxidant enzymes, and thus more susceptible to carcinogenesis than wild-type mice. 65 Among the cytoprotective proteins, HO-1 is induced by a wide array of noxious stimuli and exhibits high antioxidant activity. 66,67 Overexpression of ROS importantly contributes to TPA-stimulated JB6 P+ cell transformation. 68 In contrast, induction of HO-1 attenuates the TPA-induced ROS production and tumor promotion. As an example, zerumbone has been demonstrated to suppress TPA-induced ROS formation as well as cell transformation through the induction of Nrf2-mediated HO-1 enzymes in JB6 P+ cells. 69 In this study, we also found that RGO increases the Nrf2-mediated HO-1 expression; thereby blocking the ROS overproduction in the TPA-treated JB6 P+ cells.
In conclusion, our findings show that RGO exhibits chemopreventive properties in the TPA-stimulated JB6 P+ cell transformation. RGO pretreatment resulted in the suppression of AP-1 and NF-κB phosphorylation as well as nuclear translocation of c-Jun and p65. The TPA-induced expression of proteins involved in inflammation, proliferation, apoptosis, and angiogenesis, such as COX-2, Bcl-2, cyclin D1, and cyclin E, were also attenuated by RGO. RGO suppressed the intracellular signaling transduction pathways, including ERK, p90RSK2, JNK, and GSK-3β, involving in the TPA-induced tumor promotion. Furthermore, RGO enhanced the antioxidant defense system in the JB6 P+ cells through upregulation of Nrf2-mediated antioxidant enzyme HO-1, thereby preventing TPA-induced ROS overproduction. Overall, RGO could be a potent chemopreventive agent for the prevention of tumor promotion in skin.
Footnotes
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (NRF-2014R1A2A1A11050006).
Author Disclosure Statement
No competing financial interests exist.