
Abstract
Evidence indicates that indirect inhibitory regulation of glutamatergic transmission, via reducing glutamate release, may induce neuroprotection. The present work was designed to examine whether allicin, a major component of garlic with neuroprotective effects, affected the release of glutamate evoked by 4-aminopyridine in rat cerebrocortical nerve terminals (synaptosomes). Allicin caused a potent inhibition on the release of glutamate evoked by 4-aminopyridine, and this inhibitory effect was abolished in the presence of Ca2+-free medium and vesicular transporter inhibitor. Allicin decreased the 4-aminopyridine-evoked elevation of intrasynaptosomal Ca2+ levels, but had no effect on the synaptosomal plasma membrane potential. The allicin-mediated inhibition of glutamate release was prevented by the N- and P/Q-type channel blocker and the protein kinase C (PKC) inhibitor, but was not affected by the intracellular Ca2+-release inhibitors, mitogen-activated protein kinase inhibitor, and protein kinase A inhibitor. Western blotting data also showed that allicin significantly reduced the phosphorylation of PKC. Together, these data indicate that in rat cerebrocortical nerve terminals, allicin depresses glutamate release and appears to decrease N- and P/Q-type Ca2+ channel and PKC activity.
Introduction
Glutamate is a physiologically important excitatory neurotransmitter in the mammalian brain. Increased amounts of glutamate cause excessive stimulation of glutamate receptors and leads to neuronal death. 1 This phenomenon is well known as excitotoxicity, which has been implicated in several neuronal diseases, such as epilepsy, ischemia, stroke, trauma, or neurodegenerative diseases. 2 Animal studies have confirmed neuroprotective effects of the glutamate receptor antagonists in the central nervous system, however, these agents have been unsuccessful in clinical trials because of many adverse effects, such as ataxia, psychosis, and cognitive dysfunction. 3,4 Therefore, a more promising neuroprotective strategy for treating brain disorders may be through indirect inhibitory modulation of glutamatergic transmission, such as reducing glutamate release. 5 –8
Recently, there is an emerging trend to search for natural resources to combat against brain diseases. Many studies have reported the protective effects of various natural products against excitotoxicity models. 9 –11 Allicin is an active constituent of garlic and possesses antioxidative, anti-inflammatory, antimicrobial, anticancer, cardiovascular protective, and immunomodulatory properties. 12,13 Allicin also presents neuroprotective properties and has been investigated in various animal models with neurological disorders, including stroke, traumatic brain injury, and Alzheimer's disorder. 14 –18 However, the mechanism involved in the neuroprotective effects of allicin in the brain is not adequately understood.
Given the involvement of excessive glutamate-mediated neurotoxicity in brain injury, this work aims to study the effect and possible mechanisms of allicin on glutamate release by using rat cerebral cortex nerve terminals (synaptosomes), a model particularly suited to the evaluation of presynaptic effects on neurotransmitter release. 19 To our knowledge, a direct effect of allicin on glutamate release in synaptosome preparations has not yet been studied, and hence this will be the first report in this field.
Materials and Methods
Chemicals
DL-threo-β-benzyl-oxyaspartate (DL-TBOA), Bafilomycin A1, dantrolene, GP37157, bisindolylmaleimide I (GF109203X), 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059), and N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H89) were purchased from Tocris Cookson (Bristol, United Kingdom). 3,3′-dipropylthiadicarbocyanine iodide [DiSC3(5)] was obtained from Invitrogen (Carlsbad, CA, USA). ω-Conotoxin MVIIC was purchased from Alomone Labs (Jerusalem, Israel). Fura-2-acetoxymethyl (Fura-2-AM) ester was obtained from Life Technologies (Bengaluru, India). Allicin, 4-aminopyridine, and all other reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Experimental animals
Male Sprague-Dawley rats (2-month-old; BioLASCO, Inc., Taiwan) were used throughout the study. Animals were sacrificed by rapid decapitation. All experiments with rats were conducted according to the guidelines established by the Institutional Animal Care and Utilization Committee at Fu Jen Catholic University.
Preparation of synaptosomes
Synaptosomes from rat cerebral cortex were isolated as described previously. 20,21 In brief, the tissue was homogenized in 0.32 M sucrose; the homogenate was centrifuged for 2 min at 3000 g, and the supernatant (2 mL) was placed on a 3 mL Percoll discontinuous gradient (3%, 10%, and 23% in Tris-buffered sucrose). After centrifugation at 32,500 g for 7 min, the synaptosomes were collected from the 10% and 23% Percoll bands, and they were resuspended in hydroxyethyl piperazine ethanesulfonic acid (HEPES) buffer medium having the following composition (mM): NaCl, 140; KCl, 5; NaHCO3, 5; MgCl2·6H2O, 1; Na2HPO4, 1.2; glucose,10; HEPES, 10; and pH 7.4.
Glutamate release experiment
The release of glutamate was measured using a continuous fluorimetric assay, based on the reduction of NADP+ catalyzed by glutamate dehydrogenase in the presence of glutamate. 22,23 In short, synaptosomes (0.5 mg) were incubated for 5 min at 37°C in HEPES buffer medium (HBM) containing bovine serum albumin (BSA; 16 μM) in a PerkinElmer LS-55 spectrofluorimeter (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA). After a further 5 min incubation, NADP+ (1 mM), glutamate dehydrogenase (50 units/mL) and CaCl2 (1.2 mM) were added, and the fluorescence of nicotinamide adenine dinucleotide phosphate (NADPH) was measured, at excitation and emission wavelengths of 340 and 460 nm, respectively. Glutamate release was quantitated by the addition of 5 nmol glutamate at the end of each run, which acted as an internal standard. The data were obtained at 2-sec intervals.
Intraterminal Ca2+ concentration
Intraterminal Ca2+ concentration ([Ca2+]i) was assayed with Fura-2 as described previously. 24 Synaptosomes were preincubated in HBM containing Fura-2-AM (5 μM), CaCl2 (0.1 mM), and BSA (16 μM) for 30 min at 37°C. Then the synaptosomes were centrifuged for 1 min at 10,000 g and resuspended in HBM containing BSA (16 μM) and CaCl2 (1.2 mM) at 37°C. Fura-2-Ca fluorescence was assayed under continuous stirring, at the excitation wavelengths of 340 and 380 nm and emission wavelength of 505 nm and data accumulated at 5-sec intervals. The [Ca2+]i was calculated with the equations described previously. 25
Membrane potential measurement using DiSC3(5)
Synaptosomes in HBM containing BSA were preincubated for 30 min at 37°C. After this time, an aliquot (2 mL) was transferred to a stirred cuvette containing DiSC3(5) (5 μM) and CaCl2 (1.2 mM). After 1 min incubation, DiSC3(5) fluorescence was assayed at excitation and emission wavelengths of 646 and 674 nm, respectively, and data accumulated at 2-sec intervals. 26
Western blotting
Synaptosomes were lysed in ice-cold lysis buffer and quantified for protein content. Samples (20 μg) were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membrane. Membranes were incubated at room temperature for 1 h in Tris-buffered saline containing 5% skimmed milk and then probed with rabbit monoclonal anti-phospho-protein kinase C (PKC) (1:2000; Cell Signaling Technology, MA, USA) and anti-β-actin (1:5000; Cell Signaling Technology), overnight at 4°C. After extensive washes in Tris-buffered saline, membranes were incubated for 1 h at room temperature with appropriate peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:5000; Santa Cruz, CA, USA). Immunoblots were visualized with an enhanced chemiluminescence (Amersham, Buckinghamshire, United Kingdom) plus western blot detection system (Synoptics, Cambridge, United Kingdom).
Data analysis
Data are expressed as the mean ± standard error of the mean (SEM). Statistical significance was determined by unpaired Student's two-tailed t-tests and two-way analysis of variance with Scheffe's post hoc test. A P-value <.05 was considered statistically significant.
Results
Figure 1A shows that 4-aminopyridine triggered a glutamate release of 6.9 ± 0.2 nmol/mg/5 min in the cerebrocortical synaptosomes incubated in the presence of CaCl2 (1.2 mM). Preincubation with 10 μM allicin before 4-aminopyridine addition reduced glutamate release to 2.8 ± 0.1 nmol/mg/5 min [t(18) = 22.7, P < .001; 55.8% ± 1.5% inhibition; Fig. 1B]. Allicin had no effect on the basal release of glutamate (P > .05). The action of allicin was dose-dependent; the maximum inhibition was seen when allicin was applied at 10 μM and the half-maximal inhibitory concentration (IC50) was 6 μM (Fig. 1B).

Allicin inhibits 4-aminopyridine-evoked glutamate release from rat cerebrocortical synaptosomes.
Figure 2A shows that the evoked glutamate release was decreased in the presence of calcium-free solution that contained 300 μM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) [t(8) = 25.5, P < .001]. In the absence of extracellular calcium, the allicin (10 μM)-mediated inhibition of glutamate release was abolished [F(1.17) = 233.2, P < .001; Fig. 2A, B]. DL-TBOA (10 μM), an excitatory amino acid transporter blocker, increased 4-aminopyridine-induced glutamate release [t(9) = −8.9, P < .001] and failed to affect the inhibitory effect of allicin on evoked glutamate release [F(1.19) = 2.83, P = .11; Fig. 2A]. In the presence of DL-TBOA, allicin (10 μM) continued to significantly suppress 4-aminopyridine-induced release of glutamate (P < .05; Fig. 2B). In addition, the vesicular transporter inhibitor bafilomycin A1 (0.1 μM) reduced the glutamate release evoked by 4-aminopyridine [t(8) = 21.4, P < .001] and abolished the inhibitory effect of allicin [F(1.19) = 167.1, P < .001; Fig. 2A]. On average, in the presence of bafilomycin A1, allicin produced a 4.0% ± 7.4% inhibition, which was significantly different from the inhibition produced by allicin alone [55.8% ± 1.5%; t(9) = 6.7, P < .05; Fig. 2B]. These data indicate that allicin only inhibits the Ca2+-dependent release of glutamate evoked by 4-aminopyridine.
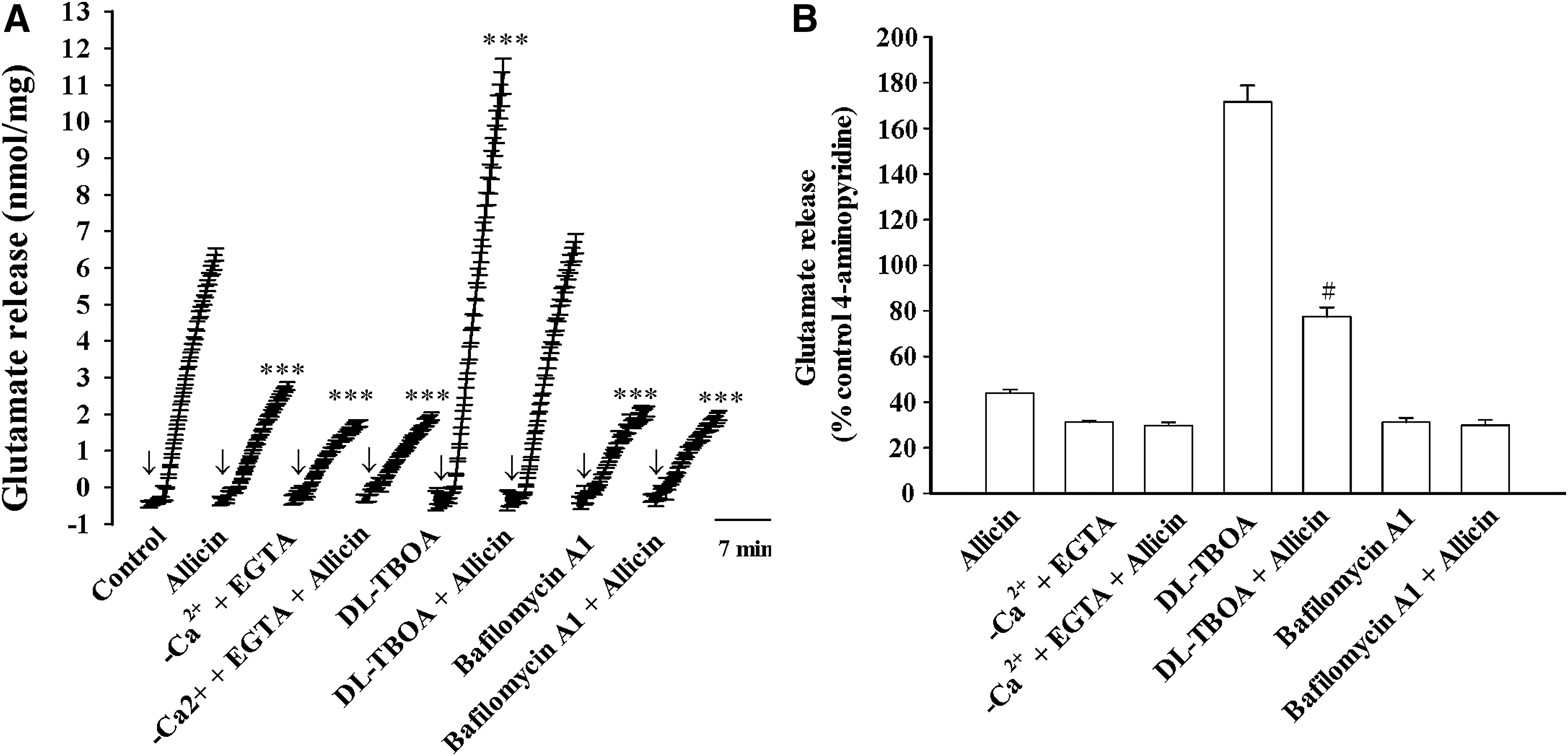
Effect of external calcium omission, the glutamate transporter blocker DL-TBOA, and the vesicular transporter inhibitor bafilomycin A1 on the allicin-mediated inhibition of 4-aminopyridine-evoked glutamate release.
Table 1 shows the effects of allicin on the [Ca2+]i and synaptosomal membrane potential. The depolarization of synaptosomes due to 4-aminopyridine (1 mM) caused a rise in [Ca2+]i to a plateau level. Preincubation with 10 μM allicin did not alter the basal [Ca2+]i, but it decreased the 4-aminopyridine-evoked rise in [Ca2+]i [t(8) = 8.1, P < .001]. In addition, 4-aminopyridine caused an increase in DiSC3(5) fluorescence. The presence of 10 μM allicin did not significantly affect the plasma membrane potential either under resting conditions or 4-aminopyridine stimulation [t(8) = 0.67, P > .05] (Table 1). These results suggest that the observed inhibition of the elevation of [Ca2+]i by allicin was most likely due to a direct reduction of Ca2+ influx through voltage-dependent Ca2+ channels, which consequently reduced glutamate release.
Effect of Allicin on Intrasynaptosomal Ca2+ Concentration ([Ca2+]i) and Synaptosomal Membrane Potential and in Rat Cerebrocortical Synaptosomes
P < .001 versus the control group; # P < .001 versus the 4-aminopyridine group.
Figure 3 assesses the role of N- and P/Q-type Ca2+ channels, which support glutamate release from synaptosomes. 27,28 The glutamate release evoked by 4-aminopyridine (1 mM) was significantly decreased by 4 μM ω-conotoxin MVIIC, an N- and P/Q-type Ca2+ channel blocker [t(8) = 21.7, P < .001]. Notably, in the presence of ω-conotoxin MVIIC, the inhibitory effect of 10 μM allicin on the glutamate release evoked by 4-aminopyridine was abolished [F(1.17) = 13.3, P < .001; Fig. 3A, B]. We also examined the inhibitory effect of allicin in synaptosome pretreated with the intracellular Ca2+-release inhibitors dantrolene (100 μM) and CGP37157 (100 μM). Figure 3A shows that the two inhibitors significantly affected the evoked glutamate release (dantrolene, t(8) = 2.6, P < .001; CGP37157, t(8) = 8.7, P < .001]. However, dantrolene or CGP37157 was not able to influence the allicin-mediated inhibition of 4-aminopyridine (4-AP) evoked glutamate release [dantrolene, F(1.17) = 13.3, P > .05; CGP37157, F(1.17) = 4.3; P > .05; Fig. 3A]. In the presence of dantrolene or CGP37157, allicin induced a statistically significant inhibition (P < .05; Fig. 3B). Therefore, a reduction of Ca2+ entry through N- and P/Q-type Ca2+ channels is involved in the inhibitory action of allicin on evoked glutamate release
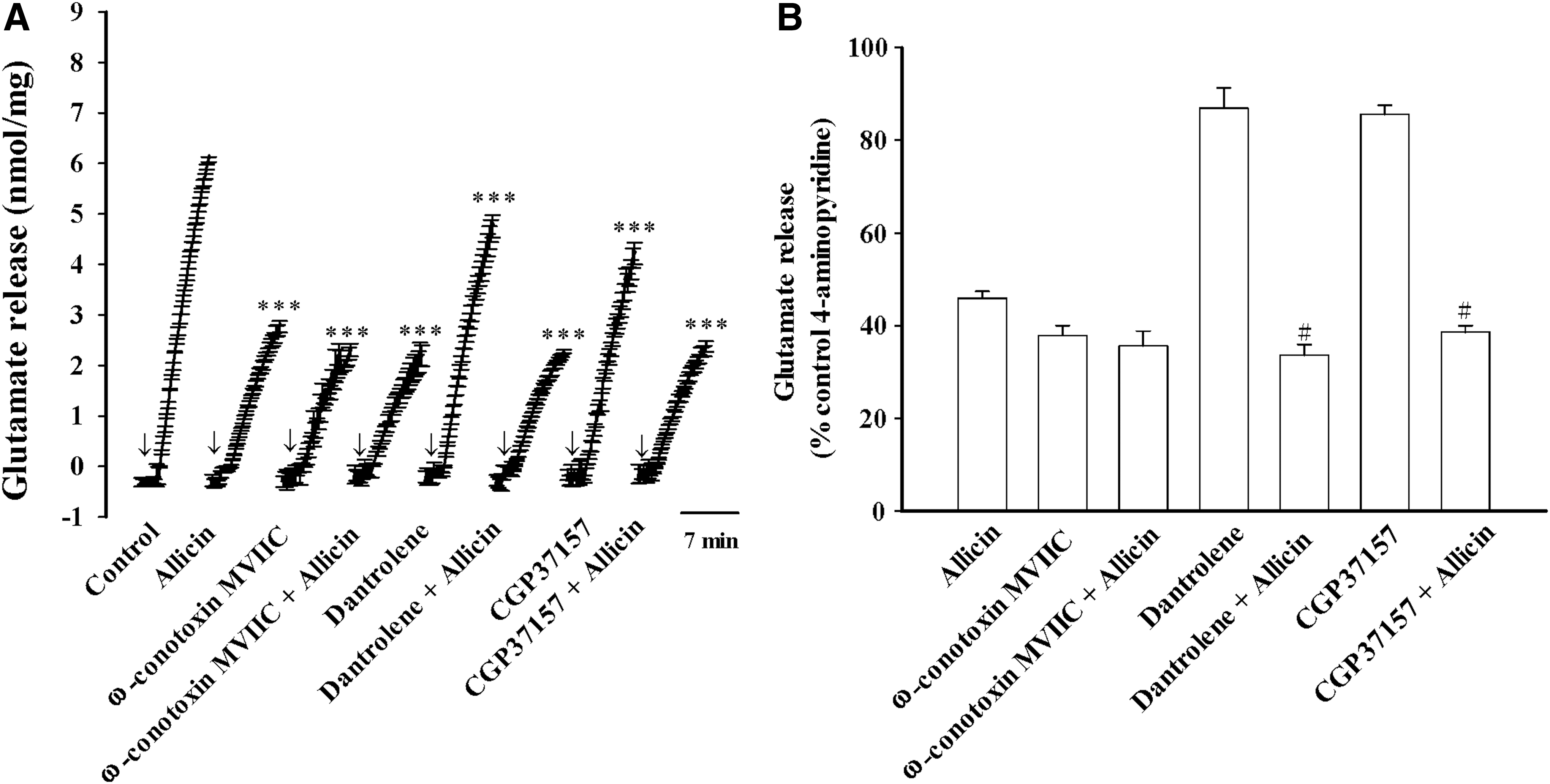
Effects of N- and P/Q-type calcium channel blocker ω-conotoxin MIIVC or intracellular Ca2+ release inhibitors dantrolene and CGP37157 on the allicin-mediated inhibition of 4-aminopyridine-evoked glutamate release.
Figure 4 shows that the evoked glutamate release was reduced by the PKC inhibitor GF109203X (10 μM) [t(8) = 39.9, P < .001], mitogen-activated protein kinase (MAPK) inhibitor PD98059 (10 μM) [t(8) = 15.1, P < .001], and protein kinase A (PKA) inhibitor H89 (100 μM) [t(8) = 7.2, P < .001]. PD98059 or H89 failed to affect the ability of allicin (10 μM) to inhibit glutamate release [PD98059, F(1.16) = 3.9, P > .05; H89, F(1.16) = 10.3; P > .05; Fig. 4A]. In the presence of PD98059 or H89, allicin produced a statistically significant inhibition (P < .05; Fig. 4B). By contrast, allicin (10 μM) only reduced glutamate release by an additional 8.2% ± 6.8% in synaptosome pretreated with GF109203X [F(1.16) = 216.8; P < .001], thus indicating a significantly higher reduction in release than that observed for the 59.5% ± 2.7% inhibition produced by allicin alone (P < .05; Fig. 4A, B). In addition, the levels of phosphorylated PKC in synaptosomes were assessed with western blotting (Fig. 5). Increases in the phosphorylation of PKC was observed after 4-aminopyridine (1 mM) addition [control, 102.8% ± 11.4%; 4-aminopyridine, 218.6% ± 14.5%; t(8) = −6.3, P < .001]. This 4-aminopyridine-increased phosphorylation of PKC was also markedly reduced after exposure to 10 μM allicin (67.9% ± 14.5%; P < .05; Fig. 5). Allicin did not significantly affect the phosphorylation of PKC in the control conditions [77.5% ± 12.9%; t(8) = 1.5, P > .05].
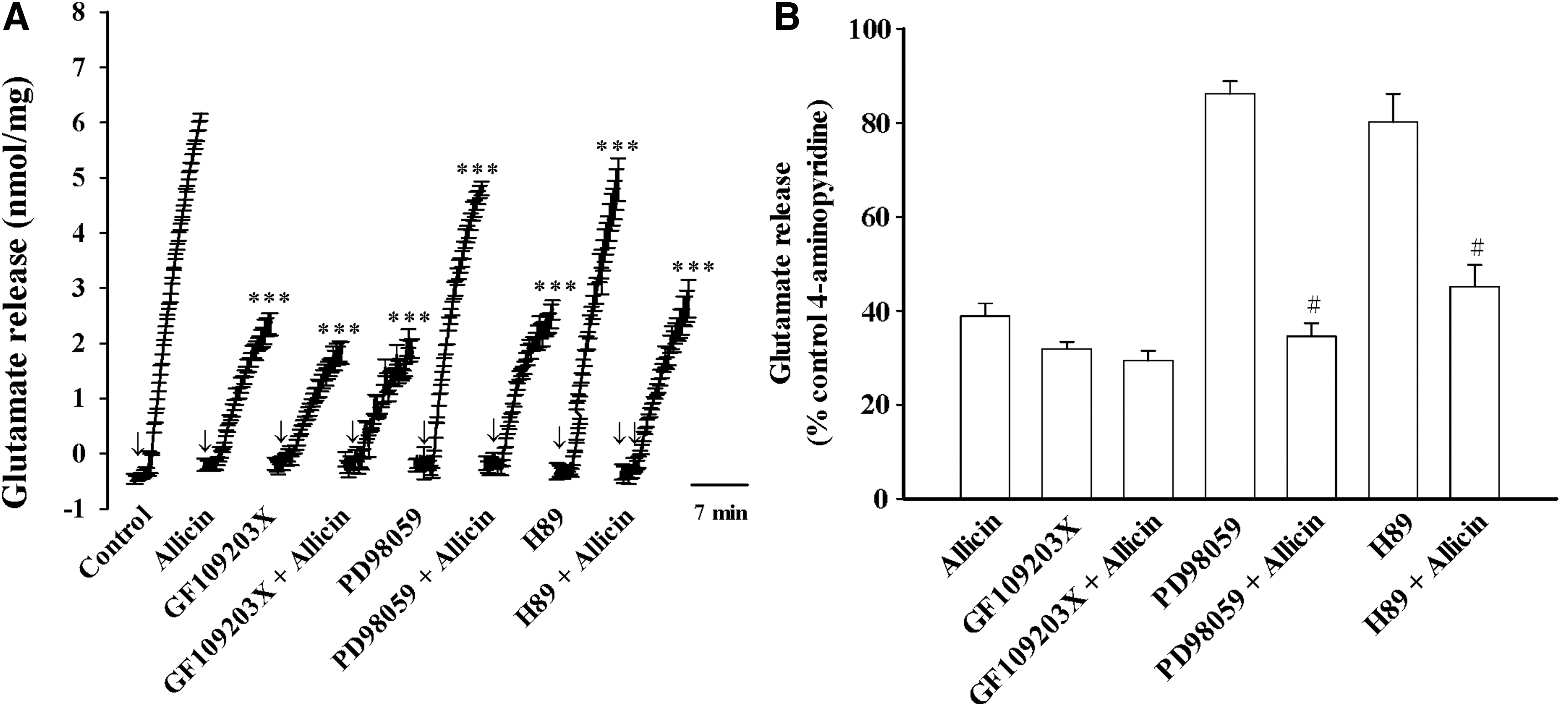
Effects of the PKC inhibitor GF109203X, MAPK inhibitor PD98059 and PKA inhibitor H89 on the allicin-mediated inhibition of 4-aminopyridine-evoked glutamate release.

Effect of allicin on the activation of PKC. The expression levels of p-PKC and PKC in synaptosomes were determined by western blotting. Data are mean ± SEM. (n = 5). ***P < .001 compared with control; # P < .05 compared with 4-aminopyridine alone.
Discussion
It is well known that excitotoxicity caused by high concentrations of glutamate in the brain has been implicated in the pathophysiology of many brain diseases. 1 Moreover, several studies have reported that reduced brain glutamate level is an important strategy for neuroprotective actions. 29,30 Allicin is a major component of garlic, which is a widespread dietary component without any toxic side effects. 12 Numerous studies have shown that allicin has a protective effect on the brain. 14 –17,31 However, a direct effect of allicin on glutamate release in isolated nerve terminals (synaptosomes) has not yet been demonstrated. Thus, in this study, the effect and probable mechanisms by which allicin affects glutamate release were evaluated using synaptosomes isolated from the rat cerebral cortex. We observed that allicin depressed the 4-aminopyridine-evoked Ca2+-dependent glutamate release, and this effect was concentration-dependent, with an IC50 value derived from a dose–response curve of ∼6 μM. Thus, we suggested that the neuroprotective effect of allicin in the brain is at least partially attributable to its glutamate release-inhibiting effect. Liu et al. reported that allicin attenuates glutamate-induced neurotoxicity in primary cultured spinal cord neurons, 32 which supported our hypothesis.
The mechanism by which allicin inhibits the evoked glutamate release in rat cerebrocortical synaptosomes is not fully understood, but it probably involves a reduction of presynaptic Ca2+ entry without any effect on synaptosomal excitability. Indeed, we observed no effect of allicin on 4-aminopyridine-evoked membrane depolarization measured with a dye of DiSC3(5). This suggests that the inhibition of glutamate release by allicin is not the result of an indirect effect via attenuation of membrane potential and thus synaptosomal excitability. Moreover, allicin failed to affect the evoked glutamate release in the absence of extracellular Ca2+, suggesting that the inhibition of glutamate release by allicin required an influx of Ca2+ from the extracellular milieu. Using Fura-2, the present work has observed that allicin significantly inhibited the evoked rise in [Ca2+]i. The inhibitory action of allicin on the evoked glutamate release was decreased from 55% to 6% after exposure to the N- and P/Q-type Ca2+ channel inhibitor, but it was not affected by the intracellular Ca2+ release inhibitors. These data imply that the observed inhibitory effect of allicin on the evoked glutamate release is attributable to a decrease in extracellular Ca2+ entry via N- and P/Q-type Ca2+ channels that are supported for glutamate release from synaptosomes. 27,28 Under conditions in which the N- and P/Q-type Ca2+ channels have been inhibited, however, total blockage of allicin action was not observed, probably due to the participation of other presynaptic pathways. Although our data indicate that allicin depresses evoked glutamate release by decreasing Ca2+ entry through N- and P/Q-type Ca2+ channels in rat cerebrocortical synaptosomes, a central question that we have not addressed in this study is, how does allicin affect these presynaptic Ca2+ channel activity? In this respect, we hypothesize that allicin inhibits presynaptic voltage-dependent Ca2+ channels either via a direct effect on Ca2+ channel function or rather indirectly via the regulation of protein kinases and consequently affected the phosphorylation of Ca2+ channels.
In the present work, we also discovered that the inhibitory effect of allicin on the release of glutamate evoked by 4-aminopyridine was efficiently blocked by a PKC inhibitor, and not by a MAPK inhibitor and a PKA inhibitor, thus indicating the involvement of PKC suppression. This conclusion was further supported by our immunoblotting data, which showed that allicin reduced the 4-aminopyridine-evoked phosphorylation of PKC in rat cerebrocortical synaptosomes. PKC is known to be present in central nervous system glutamatergic nerve endings, where they enhance the release of glutamate through several different mechanisms, including inhibiting K+ channels, increasing Ca2+ channels, and augmenting the release machinery. 33 –35 Thus, it seems that allicin elicits a reduction of Ca2+ entry through nerve terminal N- and P/Q-type Ca2+ channels, which subsequently suppresses the PKC activation to cause a decrease in evoked glutamate release. In addition to voltage-dependent Ca2+ channels, however, it has been shown that depolarization causes the reversal of the Na+/Ca2+ exchanger, leading to intracellular Ca 2 increase and glutamate release. 36 Whether allicin acts by decreasing the influx of Ca2+ through the reversal of the Na+/Ca2+ exchangers remains to be explored.
In conclusion, findings from our current study revealed that allicin, an active substance of garlic, inhibits evoked glutamate release in rat cerebrocortical nerve terminals. This phenomenon is dependent on suppression of Ca2+ influx and PKC activity. On the basis of our findings, we would suggest that allicin is a promising candidate for preventing and treating glutamate-induced neurological disorders.
Footnotes
Acknowledgment
This study was supported by the Far-Eastern Memorial Hospital (106-FEMH-FJU-03).
Author Disclosure Statement
No competing financial interests exist.