
Abstract
Oxidative stress is initiated by reactive oxygen species, the primary factor in many chronic diseases. Moringa oleifera possesses strong antioxidant properties due to the presence of various phytochemicals. In this study, we investigated the effect of M. oleifera leaf extract on markers of oxidative stress in HL60 cells exposed to oxidative stress. HL60 cells were incubated with different concentrations of M. oleifera leaf extract, and cells were harvested for viability assays on days 1, 2, and 3. Antioxidant indexes (malondialdehyde, reduced glutathione, superoxide dismutase, and catalase) were measured on days 1, 2, and 3. Supplementation with the moringa leaf extract at all concentrations resulted in significant reductions in lipid peroxidation in cells that were or were not incubated in an environment with excess oxidative stress. The most significant reduction in this parameter occurred after 24 h of incubation. The results show that reductions seen in this parameter may be due to the modulation of the endogenous antioxidant defense system by extract supplementation. Cell viability was also improved in cells incubated in moringa leaf extract at concentrations of 800 and 1000 μg/mL. This finding, however, did not corroborate with lipid peroxidation results at 1000 μg/mL extract supplementation. Further investigations are needed to clarify the underlying mechanism responsible for increased cell viability at this concentration. We can, therefore, conclude that the moringa leaf extract offered added protection from oxidative stress within the first 24 h, as well as increasing cell viability at certain concentrations.
INTRODUCTION
Oxidative stress may be initiated by reactive oxygen species (ROS) and is thought to contribute to cellular aging, mutagenesis, carcinogenesis, and other metabolic diseases, through various mechanisms including destabilization of membranes, DNA and protein damage, and oxidation of low-density lipoprotein. 1,2 Antioxidants provide protection against oxidative stress by suppression of various cellular activities that lead to ROS formation. The leaves of a tropical plant Moringa oleifera are thought to possess antioxidant properties that may protect cells against free radical damage.
Moringa oleifera Lam is thought to be the most popular species of the Moringaceace family and is thought to be one of the world's most dynamic plants. 3 It is widely grown and utilized for its nutraceutical properties in diverse locations. 4 Interestingly, different parts of the plant are utilized for cleaning purposes, as a biopesticide and in the cosmetic industry. 5,6 Studies show that moringa seeds are useful as biosorbents for the removal of heavy metal from aqueous medium and are the best known natural coagulants widely utilized in the food industry. 7,8
It is theorized that nutraceutical properties of many crude plant extracts are in part due to their ability to reduce oxidative stress brought on by environmental factors. Oxidative stress initiated by ROS is identified as a primary contributor to proliferation of malignant cells. 9 Numerous plant extracts can treat a wide variety of maladies, and it is with this in mind that we explore the benefits of the methanolic extracts of M. oleifera leaves against human myeloproliferative leukemia (HL60) cells subjected to oxidative stress.
MATERIALS AND METHODS
Collection of plant materials
Fresh samples of M. oleifera leaves were collected on the campus of the University of the West Indies, Mona, and in the community of Mona, Kingston, Jamaica. Voucher specimens of the leaves of M. oleifera (No. 36475) were deposited in the herbarium of the department of botany at the University of the West Indies, Mona. Samples were air-dried in the laboratory for 2 weeks. Leaves were then milled with a Waring Blender and stored in zip-lock bags at 4°C for future use.
Preparation of solvent extract
Crude extract preparation was carried out as described by Dehshahri and others. 10 Five hundred grams of powdered samples was separately defatted with n-hexane and then extracted with 500 mL methanol–water (80:20) at room temperature by maceration with occasional stirring. Samples were then covered with aluminum foil and placed in the dark for 24 h. At the end of each 24-h period, the solvent was decanted. This was repeated two times and the extracts were combined. The combined extracts were concentrated to dryness with a rotary evaporator (Buchi, Switzerland; temperature: 50°C; pressure 175 mbar) and vacuum oven at temperatures between 40°C to 50°C. The crude extract was then preserved at 4°C in airtight bottles until required for use. Final concentration of the extract was 200 mg/mL. The extract was dissolved in 1.0 mL of dimethyl sulfoxide (DMSO) and filtered through a 0.22 μM filter. From this was prepared crude extract concentrations of 100, 200, 300, 400, 500, and 600 μg/mL. The sterile extracts were prepared and used in a cell culture hood under aseptic conditions.
Preparation of cultured HL60 cells
The human promyelocytic leukemia cell line HL60 (passage <100) was obtained from American Type Culture Collection (ATCC, Rockville, MD, USA) and maintained in RPMI-1640 medium and Dulbecco's modified Eagle's medium, respectively, with 10% heat-inactivated fetal bovine serum (Corning Cellgro, Manassas, VA, USA). Media contained 2 mmol/L of glutamine, supplemented with 1% (v/v) nonessential amino acids, 1% (v/v) sodium pyruvate, 50 U/mL penicillin, and 50 mg/mL streptomycin. The cell suspension was incubated in a humidified atmosphere containing 5% carbon dioxide (CO2) at 37°C. On reaching the desired 106 cells/mL, cells were harvested according to the manufacturer's instructions (ATCC).
Preparation of hydrogen peroxide (H2O2)-induced stress HL60 cells was carried out based on methods previously described. 11,12 A volume of 0.1 mol/L H2O2 was added to fresh cell culture medium so that the final concentration of H2O2 was 50 μmol/L. HL60 cells were suspended in the prepared medium containing 50 μmol/L. HL60 cells were subsequently suspended in this medium and adjusted to the concentration of 106 cells/mL by using the trypan blue (Corning Cellgro) method. The leaf extracts were then added.
Determination of cell concentration
Trypan blue is a widely used assay for staining dead cells. After staining, cell viability was determined by counting the unstained cells. A volume of 200 μL HL60 cells was added to 300 μL of a 0.4% trypan blue solution and 500 μL phosphate buffered saline (PBS) in a test tube. The suspension was vortexed to ensure homogeneity. Ten microliters of the suspension was pipetted unto the hemocytometer and covered with a coverslip. Counting encompassed cells present in five different 1 mm squares, one center as well as four corner squares. Total number of cells was determined as follows: average cells counted per square × dilution factor × 104.
Cytotoxicity assays
Viable cells at a concentration of 1.5 × 104 were seeded in 96-well plates that were preincubated for 24 h in a humidified incubator at 37°C with 5% CO2. At 80% confluence, the medium was replaced with crude leaf extract of M. oleifera in DMSO (0.04% v/v) at concentrations of 200, 400, 600, 800, and 1000 μg/L to test for cytotoxicity. The ratio of extract to cell was 1:10. Incubation was also carried out with a positive control (20 μg/mL). After 24 h, the supernatants were removed, and the cells were washed with PBS then incubated with 10 μL of cell counting kit-8 (CCK-8) solution for 4 h in a humidified atmosphere at 37°C. The absorbance of colored formazan was measured at 450 nm by an ELISA (Technical Manual CCK-8; Dojindo, Rockville, MD, USA).
Modulatory effects of an M. oleifera leaf extract on oxidative stress in H2O2-challenged HL60 cells
Once cells were treated with both extracts at concentrations as already outlined, they were harvested for assays of catalase (CAT), superoxide dismutase (SOD), reduced glutathione (GSH), and lipid peroxidation.
Catalase
Cellular pellets (106 cells/mL) were harvested and lysed by sonication in buffer containing 50 mM potassium phosphate (pH 7.0) and 1 mM ethylenediaminetetraacetic acid (EDTA). The lysate was centrifuged at 10,000 g for 15 min at 4°C. The activity of CAT was measured in the supernatant based on methods previously described by Genet et al., 13 and Nawaz and Hasnain. 14 The reaction mixture was prepared by adding 500 μL of 0.1 M sodium phosphate buffer pH 7.0 (50 mM), 100 μL of H2O2 (10 mM), and 20 μL of the cellular supernatant treated with Triton X-100. The decrease in absorbance was measured at 240 nm for 5 min at 25°C against a blank containing all the ingredients without the supernatant. One unit of CAT is defined as the amount of enzyme required to decompose 1 μmol of H2O2.
Superoxide dismutase
The cell pellets (106 cells/mL) were lysed by sonication on a buffer [cold 20 mM HEPES buffer (pH 7.2) containing 1 mM EDTA, 210 mM mannitol, and 70 mM sucrose] (Technical Manual for SOD Assay; Cayman Chemical, Ann Arbor, MI, USA). After sonication, the lysate was centrifuged at 1500 g for 5 min at 4°C. The cellular supernatant was used for the measurement of SOD activity. One millilitre of reaction mixture was prepared by adding 500 μL of 0.1 M sodium phosphate buffer, 32 μL of 3.3 mM EDTA, 60 μL of 8.1 mM pyrogallol and appropriate amount of cellular supernatant containing 7–10 μg protein. The change in absorbance at 420 nm of the mixture was monitored for 2 min at 25°C against the blank that contains all ingredients except the supernatant. One unit of enzyme is defined as the amount of enzyme that causes half maximal inhibition of pyrogallol autoxidation. 13
Reduced glutathione
GSH was assessed by a slightly modified Ellman's method as previously described. 15 In brief, the pellets (106 cells/mL) were harvested and lysed in a hypotonic solution at 37°C for 45 min. One hundred microliters of lysate was mixed with 10% tricarboxylic acid and centrifuged at 3000 g for 15 min. Two hundred microliters of the supernatant was mixed with 100 μL Ellman's reagent (19.8 mg of 5,5’-dithiobisnitrobenzoic acid [DTNB] in 100 mL of 0.2 mol/L phosphate buffer [pH 8.0]. The absorbance of the resulting solution was read at 412 nm.
Lipid peroxidation
The pellets (106 cells/mL) were collected and suspended in ice-cold buffered saline, after which they were lysed by high power sonication at 4°C. Samples were centrifuged at 28,000 g for 5 min at 4°C and the supernatant was collected. The cellular components were quickly used for measurement of malondialdehyde (MDA) levels. The measurement of MDA was carried out by the method of Genet et al. 13 In summary, the final reaction mixture of 3 mL contained the following: 1.5 mL of 10 mM potassium phosphate buffer (pH 7.4), 0.5 mL of the cellular supernatant, 0.5 mL of 30% trichloroacetic acid, and 0.5 mL of thiobarbituric acid (0.53%). The mixture was heated for 1 h at 80°C, cooled and centrifuged for 5 min at 2,000 g at 4°C. Absorbance of the clear supernatant was measured at 532 nm against a blank. Total protein in the cellular supernatant was determined by using Stanbio Kit (Instruction Manual; Stanbio Laboratory, Boerne, TX, USA). The result is expressed in nmol of MDA formed per milligram of protein per milliliter.
Statistical analysis
Data are presented as mean ± standard error of the mean. Values were obtained from three separate experiments and each experiment included the control and five different concentrations of M. oleifera leaf extract performed in triplicates. The mean difference among different concentrations was assessed by one-way analysis of variance. Post hoc analysis was carried out using Duncan's multiple range test to ascertain significant difference among the means (P < .05).
DISCUSSION
There were no significant changes observed in viability of cells exposed to Moringa leaf extract at concentrations of 600 μg/mL or lower (Fig. 1). However, at a concentration of 800 μg/mL, the concentration of HL60 cells increased significantly compared with cells exposed to 1000 μg/mL (Fig. 1). This was despite significant reduction in lipid peroxidation in all groups. This suggests that factors other than lipid peroxidation may have been associated with the increased viability in cells observed. Nonetheless, cells exposed to moringa leaf extract concentration of 800 μg/mL or higher displayed increased viability compared with cells exposed to lower concentrations of moringa leaf extract. Of note, reductions in cell viability were observed at 1000 μg/mL compared with 800 μg/ml (Fig. 1). This indicates that the ability of moringa to positively affect cell viability is restricted to a concentration of 800 μg/mL, and above which the beneficial effects may be diminished. Moringa leaf extract at this concentration was also able to increase cell viability of HL60 cells exposed to oxidative damage to a greater extent than unexposed cells. The ability of the crude extract and its constituents to preserve cell integrity, thereby increasing their viability amid H2O2-induced oxidative damage, should be further explored.

HL60 cells incubated with varying concentrations of crude Moringa oleifera leaf extract. A comparison was made with cells exposed to oxidative damage by exposure to H2O2. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05. H2O2, hydrogen peroxide.
Cells undergo oxidative stress once affected by free radicals such as the superoxide anion. Free radicals are continuously being produced by both exogenous and endogenous sources. 16 These free radicals can attack lipid-rich structures such as lipid membranes leading to lipid peroxidation. 17 The formation of lipid peroxidation products, for example, MDA can, in turn, promulgate free radical reactions, thereby worsening the scenario. 18 Cells, however, possess an antioxidant defense system that helps to regulate intracellular concentrations of ROS by removing them. 19 CAT, glutathione peroxidase, and SOD are the three primary enzymes of the antioxidant defense system. SOD converts superoxide radicals into H2O2 and molecular oxygen. CAT and glutathione peroxidase, in turn, breaks down H2O2 to water in the case of glutathione peroxidase and water and oxygen in the case of CAT.
This study revealed significant reductions in the MDA content of the cells for a period of 72 h. In vitro studies have showed that moringa leaf extract can inhibit increase in MDA in rats exposed to toxins. 20 Reduction in lipid peroxidation levels occurred at a faster rate in cells exposed to the moringa leaf extract than in unexposed cells. The most significant reduction was observed in cells exposed to moringa leaf extract at concentrations of 600 μg/mL (cells exposed to oxidative damage) and 800 μg/mL (cells exposed to nonoxidative and oxidative damage) within the first 24 h (Fig. 2). These results were somewhat in line with SOD activities of HL60 cells incubated in extract for 24 h. This enzyme activity showed the greatest increase in cells incubated in moringa leaf extract at concentrations of 600 and 800 μg/mL and slight increases observed at 200, 400, and 1000 μg/mL (Fig. 3). SOD catalyzes the conversion of superoxide ions to H2O2, which is, in turn, broken down to release water by glutathione peroxidase, and water and oxygen by CAT.
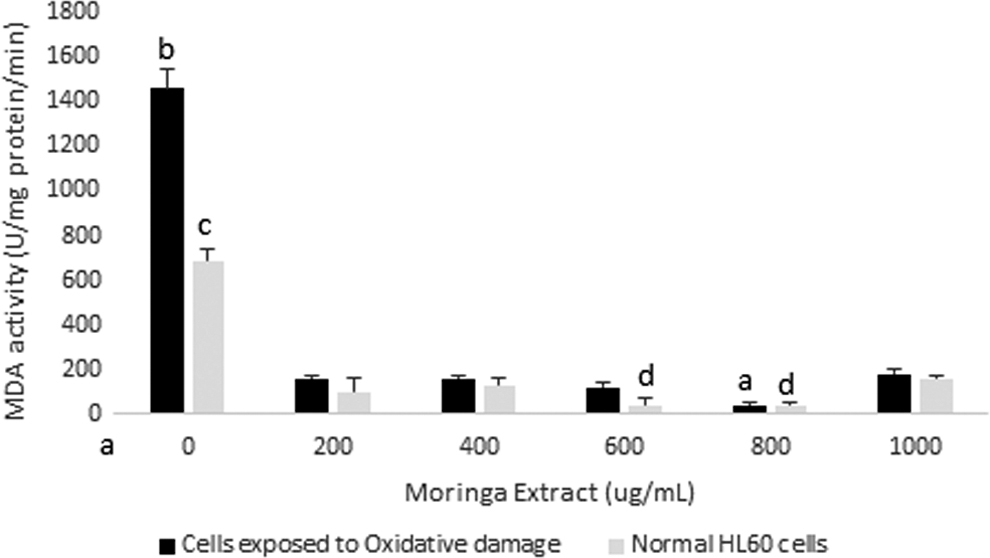
MDA activity in HL60 cells incubated with varying concentrations of crude Moringa oleifera leaf extract for 24 h. A comparison was made with cells exposed to oxidative damage by exposure to H2O2. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05. MDA, malondialdehyde.
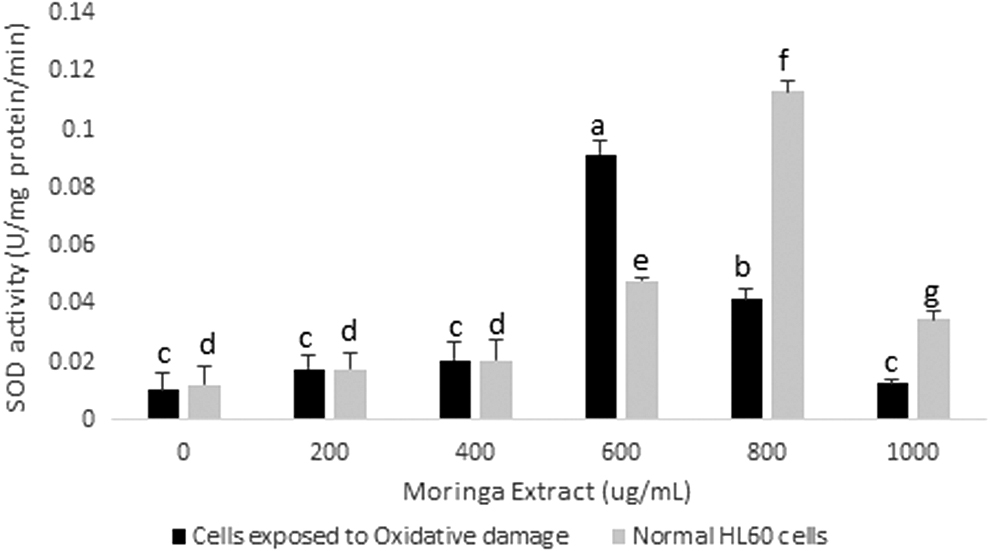
SOD activity in HL60 cells incubated with varying concentrations of crude Moringa oleifera leaf extract for 24 h. A comparison was made with HL60 cells exposed to oxidative damage by exposure to H2O2. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05. SOD, superoxide dismutase.
Significant decreases were seen in the concentrations of GSH in cells (oxi and non-oxi) incubated in 600 μg/mL moringa leaf extract, when compared with cells not incubated in the extract (Fig. 4). This suggests that the activity and/or efficiency of glutathione peroxidase may have been enhanced with the supplementation of 600 μg/mL moringa leaf extract (Fig. 5). Bermingham et al. showed that selenium supplementation from selenium-enriched foods affects glutathione peroxidase activity in various tissues, 21 whereas Gopalakrishnan et al. reported that M. oleifera leaves can be used as a source of dietary selenium. 6 Significant increase in CAT activity was also observed in cells (oxi) incubated for 24 h with extract supplementation at concentrations 200, 400, 800, and 1000 μg/mL when compared with cells incubated without extract supplementation (Fig. 6). The increase at 800 μg/mL was significantly greater than at other concentrations used in the study and could possibly be a part of the mechanism involved in the significant reduction in lipid peroxidation in cells exposed to the extract at this concentration. Based on the mentioned findings, we postulate that CAT is the main catalytic enzyme involved in the reduction of superoxide radicals in the cells incubated in 800 μg/mL.

GSH activity in HL60 cells incubated with varying concentrations of crude Moringa oleifera leaf extract for 24 h. A comparison was made with cells exposed to oxidative damage by exposure to H2O2. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05. GSH, reduced glutathione.
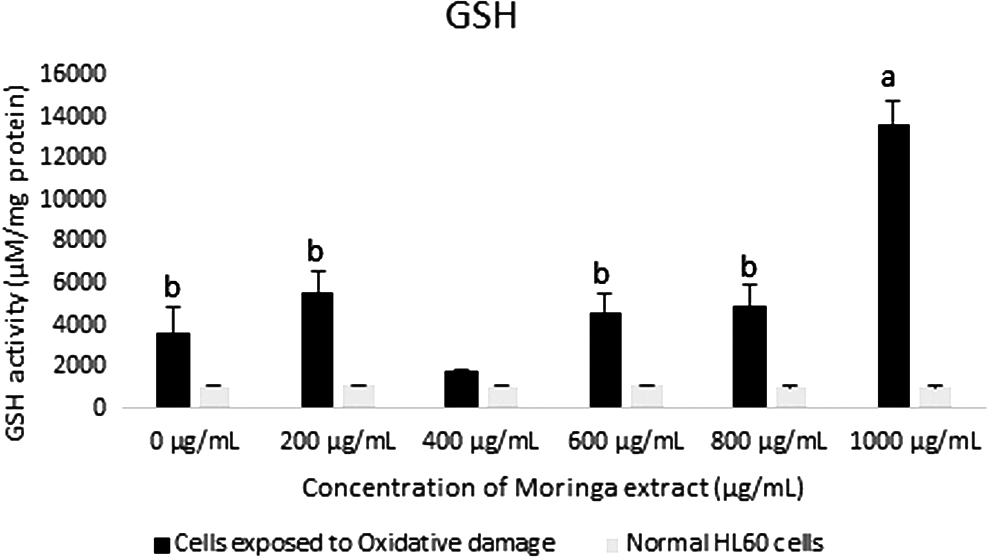
GSH activity in HL60 cells incubated with varying concentrations of Moringa oleifera leaf extract for 48 h. GSH activity was assessed in cells exposed to oxidative damage and compared with normal cells. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05.
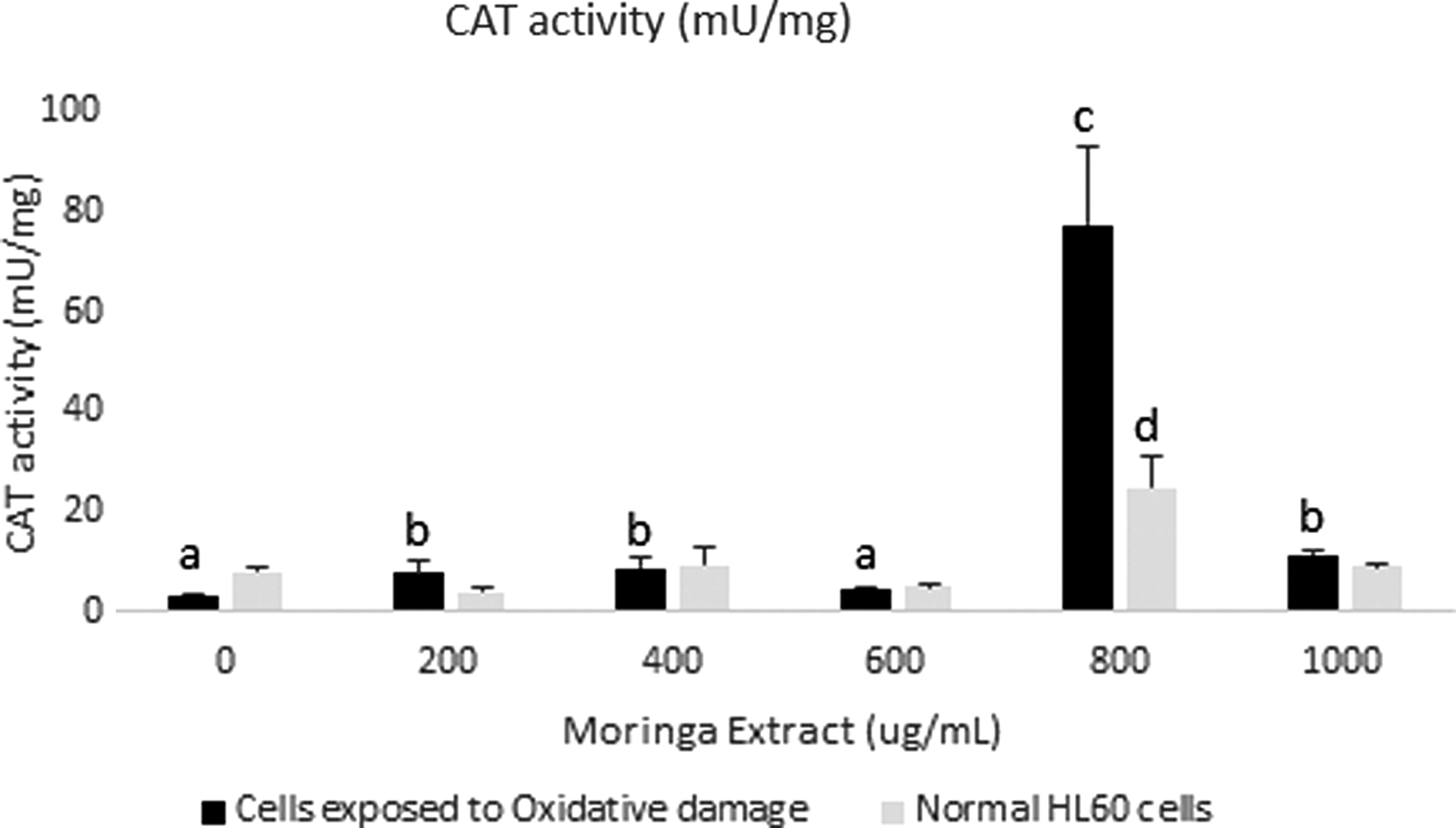
CAT activity in HL60 cells incubated with varying concentrations of crude Moringa oleifera leaf extract for 24 h. A comparison was made with cells exposed to oxidative damage by exposure to H2O2. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05. CAT, catalase.
Lipid peroxidation in cells incubated for 48 h was significantly lower than those incubated for 24 h. Cells incubated in a high oxidative stress environment for a period of 48 h had higher levels of lipid peroxidation than cells incubated in a normal environment. The extract was successful in causing significant reductions in oxidative stress in cells (oxi) growing in media containing 400, 600, 800, and 1000 μg/mL of extract compared with cells (oxi) grown without extract (Fig. 7). No significant difference was, however, observed in cells grown in medium containing 200 μg/mL extract. The significant reductions in MDA may have occurred as a result of significant increases in SOD (Fig. 8) and CAT (Fig. 9) activities, and also glutathione peroxidase activity in the case of cells incubated in 400 μg/mL of extract. In cells not exposed to high oxidative stress environments, no significant differences were observed among cells incubated with the extract, at varying concentrations, and cells incubated without extract exposure. The results suggest that under normal circumstances, the extract may not provide any additional protection after 48 h of incubation and at lower levels of lipid peroxidation.
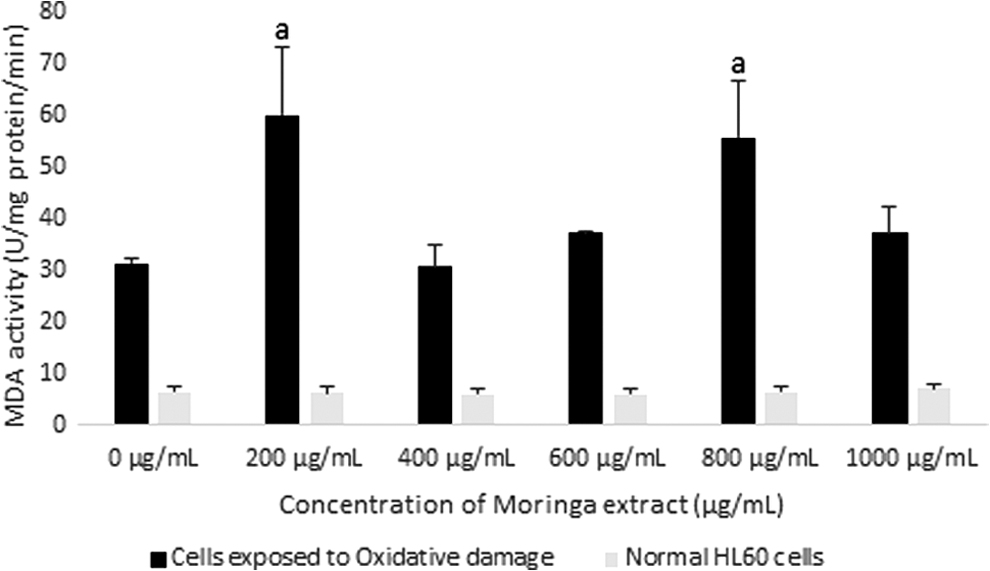
Changes in MDA activity in HL60 cells after exposure to varying concentrations of Moringa oleifera leaf extract for 48 h. A comparison of MDA activity was made between cells exposed to oxidative damage (H2O2) and normal cells. All assays were carried out in triplicates. Groups with similar subscripts are not significantly different, P < 0.05.
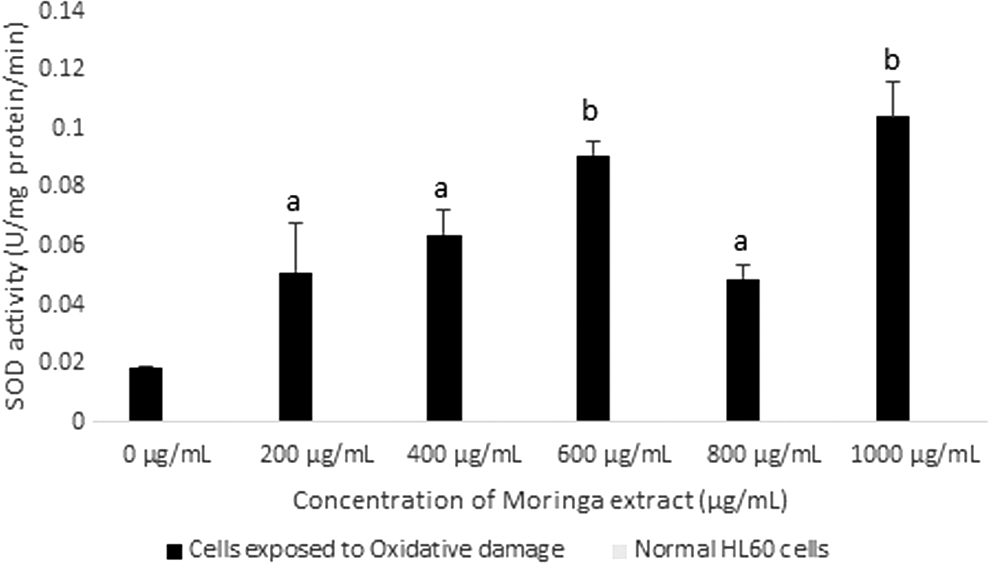
SOD activity in HL60 cells after exposure to varying concentrations of Moringa oleifera leaf extract for 48 h. A comparison of SOD activity was made between cells exposed to oxidative damage (H2O2) and normal HL60 cells. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05.
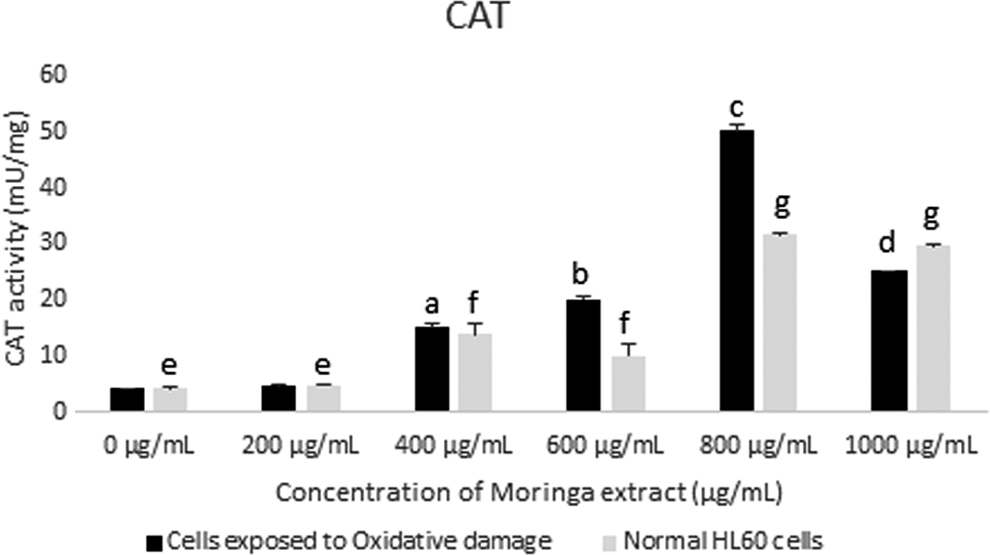
CAT activity in HL60 cells incubated with varying concentrations of Moringa oleifera leaf extract for 48 h. CAT activity was assessed in cells exposed to oxidative damage and compared with normal cells. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05.
Lipid peroxidation was observed to be lowest in all cells after 72 h of incubation (Fig. 10). No significant differences were observed between cell groups exposed to exogenous H2O2, suggesting that after 3 days of incubation, the cells were effectively able to protect themselves with or without extract supplementation. This is despite the significant increase seen in CAT and SOD activities in cells supplemented with moringa leaf extract compared with cells incubated without extract (Figs. 11 and 12). Lipid peroxidation levels in extract-supplemented cells not exposed to exogenous H2O2 were either not affected or were significantly reduced (200, 400, and 600 μg/mL extract) when compared with cells not supplemented with the extract. This suggests that extract supplementation had no negative effect on these cells and provided added protection in certain circumstances.
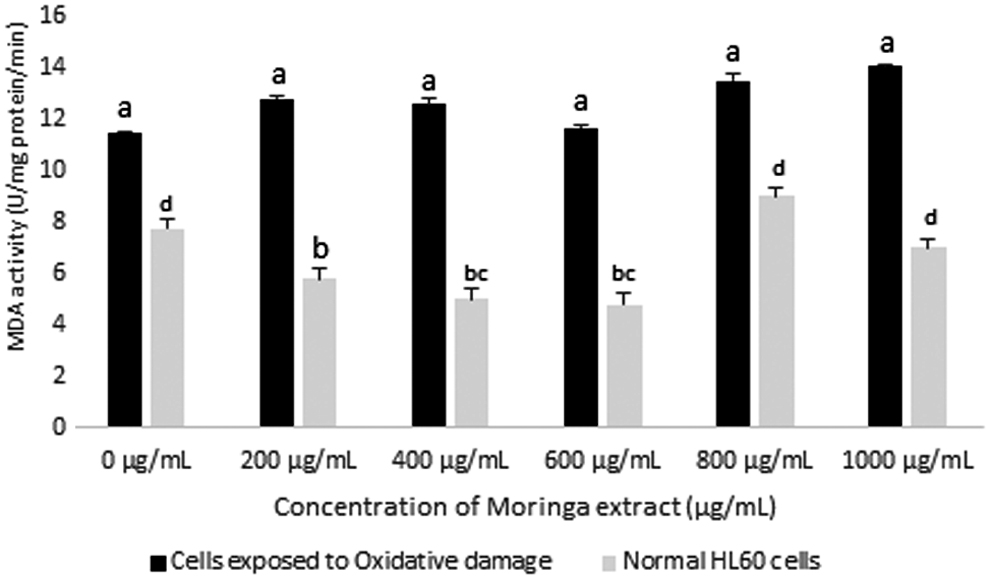
Changes in MDA activity in HL60 cells after exposure to varying concentrations of Moringa oleifera leaf extract for 72 h. A comparison of MDA activity was made between cells exposed to oxidative damage (H2O2) and normal cells. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05.
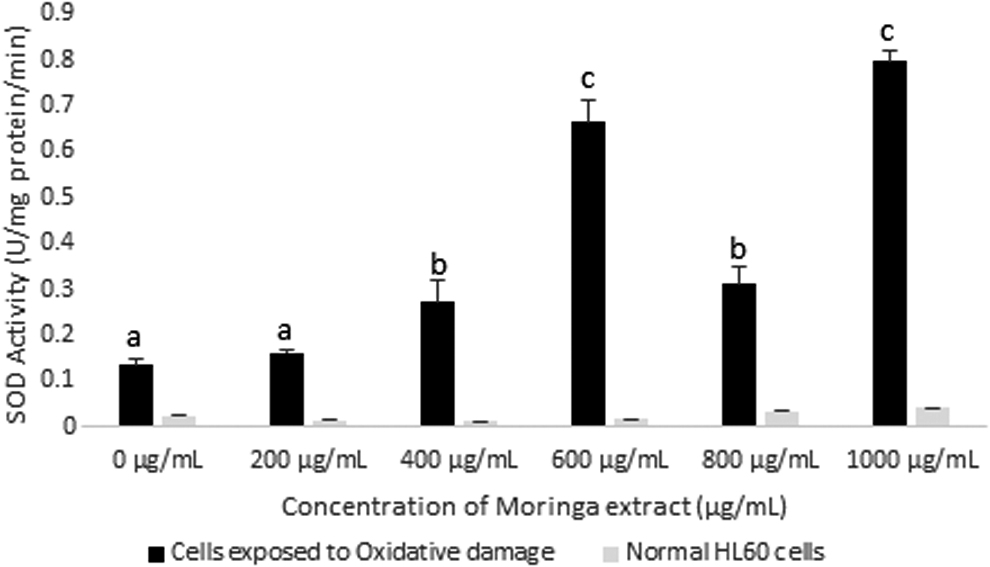
SOD activity in HL60 cells incubated with Moringa oleifera leaf extract for 72 h. All assays were carried out in triplicates. A comparison of SOD activity was made between cells exposed to oxidative damage (H2O2) and normal HL60 cells. Groups with different letter subscripts are significantly different, P < .05.

CAT activity in HL60 cells incubated with varying concentrations of Moringa oleifera leaf extract for 72 h. CAT activity was assessed in cells exposed to oxidative damage and compared with normal cells. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05.
It can, therefore, be concluded that supplementation with the moringa leaf extract at concentrations of 200 to 1000 μg/mL may have resulted in the reduction of lipid peroxidation products in normal cells and cells exposed to excess oxidative stress for 24 h. Over time, however, cells grown in both the presence and absence of excess oxidative stress were able to effectively reduce lipid peroxidation levels. In cells grown in a normal environment, moringa extract supplementation (200, 400, and 600 μg/mL) provided added protection by further reducing lipid peroxidation after being incubated for 72 h (Fig. 13). It was also revealed that extract supplementation at 800 and 1000 μg/mL resulted in significant increases in cell viability. This did not corroborate with oxidative stress parameters; hence further research is needed to clarify the underlying mechanisms involved.
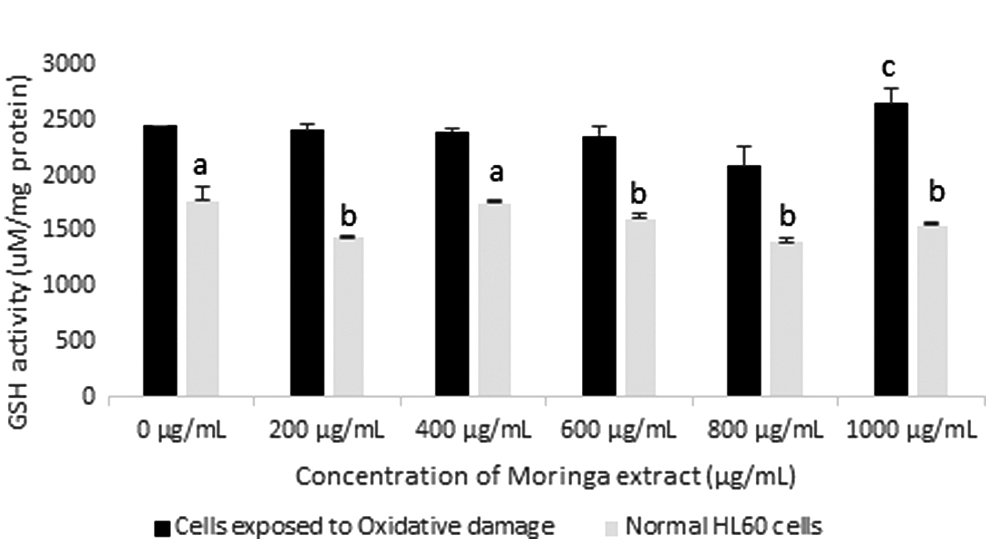
GSH activity in HL60 cells incubated with varying concentrations of Moringa oleifera leaf extract for 72 h. GSH activity was assessed in cells exposed to oxidative damage and compared with normal cells. All assays were carried out in triplicates. Groups with different letter subscripts are significantly different, P < .05.
CONCLUSION
The viability of cells supplemented with M. oleifera leaf extract was significantly improved, with the most effective dose or concentration for supplementation being 800 μg/mL. This improvement in viability was observed both in cells exposed to oxidative damage and cells that were not exposed. This improvement in viability may have been as a result of the significant reduction in oxidative stress observed in extract-supplemented cells as well as increase in the activities of enzymes of the antioxidant system. Further research is, however, needed to validate the claims made within this article and to determine the exact mechanisms associated with the increase in cell viability observed.
Footnotes
ACKNOWLEDGMENTS
We thank Dr. Dennis Bailey from the Department of Chemistry UWI Mona for sourcing plant materials for the study and also Miss Khan Nguyen at the Texas A&M University for technical guidance.
AUTHOR DISCLOSURE STATEMENT
The authors hereby disclose no conflict of interest of any kind whether financial, commercial, or personal.
FUNDING INFORMATION
No funding was received for this study.