
Abstract
The present study aimed to investigate the protective role of the flavonoid fisetin (FI) on inflammation-mediated metabolic diseases, especially tissue fibrosis and insulin resistance (IR) in high-fat diet (HFD)-induced obese mice. C57BL/6J mice were fed with normal-fat diet, HFD (40 kcal% fat), or HFD +0.02% (w/w) FI for 16 weeks. Dietary FI supplementation improved hepatic steatosis by restricting lipogenesis, while promoting lipolysis in the liver. FI also prevented adiposity via an increase in the expression of genes involved in FA oxidation and a decrease in the expression of genes involved in lipogenesis in white adipose tissue. In addition, FI increased brown adipose tissue (BAT) and skeletal muscle weights, thermogenic gene mRNA expression in BAT, and tricarboxylic acid cycle-related gene expression in skeletal muscle, which may be linked to the prevention of nonalcoholic fatty liver disease as well as adiposity. Moreover, FI supplementation decreased excessive reactive oxygen species production by increasing paraoxonase activity, adipokine dysregulation, proinflammatory cytokine production, and extracellular matrix amassment in the liver. FI supplementation ameliorated IR, in part, by normalizing pancreatic islet dysfunction, and it declined hepatic gluconeogenesis and proinflammatory responses. Taken together, the present findings indicate that FI can protect against HFD-induced inflammation-mediated disorders, including fibrosis and IR.
Introduction
Obesity is a complex disease that is closely associated with the increased risk of metabolic disorders, especially nonalcoholic fatty liver disease (NAFLD), hepatic fibrosis, insulin resistance (IR), and type II diabetes. Molecular links between obesity, NAFLD, and hepatic fibrosis may consist of extracellular matrix (ECM) accumulation and chronic inflammation within the liver, although it is not yet fully understood. 1 –3 Obesity triggers adipocyte hypertrophy, which can contribute to low-chronic inflammation and the production and accumulation of ECM, resulting in the pathogenesis of fibrosis. 4 Moreover, accumulated evidence indicates that the inflammatory reaction is complex and often entails reactive oxygen species (ROS). Oxidative stress-induced by ROS increases the proinflammatory cytokine production, and subsequently provokes the fibrogenesis signaling pathways. 5 More recently, research has been underway to prove the correlation between brown adipose tissue (BAT), skeletal muscle, and NAFLD. 6 –8 As a result, attention is focusing on BAT and skeletal muscle as target organs for treating or preventing NAFLD.
Fisetin (FI, 3,7,3′,4′-tetrahydroxyflavone) is a naturally occurring flavonoid found in fruits and vegetables, especially strawberries and blueberries. 9 FI can suppress the proinflammatory response, with strong antioxidant and antitumor effects. 10,11 FI has also been demonstrated to have antiobesity and anti-NAFLD properties. 12 –15 In high-fat diet (HFD, 60 kcal% fat)-fed C57BL/6 mice, intraperitoneal injection with FI for 10 weeks decreased body weight (BW), epididymal white adipose tissue (WAT), serum-free fatty acid (FFA) and leptin concentrations, and liver lipid droplet and hepatocyte steatosis. 13 In rat models of diet-induced obesity, FI supplementation for 8 weeks ameliorated diet-induced NAFLD by suppressing hepatic lipid accumulation, and mitigating oxidative stress. 14,15 In addition, FI given by gavage for 20 weeks, restrains HFD (60 kcal% fat)-induced hepatic inflammation and lipid deposition by downregulating metabolic disorder and excessive liver inflammation-associated TNF-α/RIPK3 axis activation. 16 However, to date, the effects of this chemical on hepatic fibrosis are not fully elucidated, and the above-mentioned research articles have provided FI using not oral supplementation but intraperitoneal injection or gavage and used unusually HFDs (60 kcal% fat), which do not reflect human dietary consumption. Moreover, little research has been done to investigate the effects of long-term oral supplementation with FI in the development of obesity and its complications. Thus, this study was designed to investigate the metabolic actions of long-term supplementation with dietary FI in HFD-induced obese mice. In particular, we focused on its role in amelioration of obesity-induced inflammation-mediated metabolic disorders, especially liver fibrosis and IR.
Materials and Methods
Experimental animals and diet
Male 4-week-old C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME, USA) were individually housed in a pathogen-free barrier facility at a room temperature (22°C ± 2°C) and maintained on a 12-h light/dark cycle. Mice were randomly assigned to a normal-fat diet (n = 13) group, a HFD (n = 13; 20% fat and 1% cholesterol) group, or HFD with 0.02% (w/w) FI (FI, n = 13; obtained from Sigma-Aldrich, St. Louis, MO, USA) group. The mice were fed each diet and water ad libitum for 16 weeks.
At the end of the diet period, the mice were sacrificed, and then blood samples, liver, WAT of epididymal, perirenal, retroperitoneum, subcutaneous, and interscapular depots, as well as interscapular BAT and skeletal muscle were isolated immediately, weighed, and stored at −70°C. The animal study protocols were approved by the Kyungpook National University Ethics Committee (Approval No. KNU 2010-4-14).
Blood analysis
The plasma FFA (Reagent1: #434-91795, Reagent2: #436-91995, NEFA Standard: #270-77000) and phospholipids (#296-63801) levels were measured using a Wako enzymatic kit (Wako Chemicals, Osaka, Japan). The plasma triglyceride (TG, #AM157S), total-cholesterol (TC, #AM202), high-density lipoprotein cholesterol (HDL-C, #AM203), glutamic oxaloacetic transaminase (GOT, #AM103-K), and glutamic pyruvic transaminase (GPT, #AM102) concentrations were determined using commercial enzymatic kits (Asan Pharm Co., Seoul, South Korea). The levels of plasma apolipoprotein A1 (ApoA1, #1433501) and B100 (ApoB100, #1433701; Eiken Chemical Co., Tokyo, Japan), and soluble CD antigen 163 (sCD163, #MBS903944) and phospholipid transfer protein (PLTP, #MBS008754) were analyzed using a commercial kit (Mybiosource, San Diego, CA, USA). The atherogenic index (AI) and HDL-C-to-TC ratio (HTR) were calculated as follows: AI = (TC − HDL-C)/HDL-C; and HTR (%) = HDL-C/TC × 100. The levels of plasma insulin, glucagon-like peptide-1 (GLP-1), leptin, adiponectin, tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), interferon γ (IFN-γ), interleukin-6 (IL-6), and plasminogen activator inhibitor-1 (PAI-1) were determined using a MILLIPLEX MAP Mouse Metabolic Hormone (GLP-1), Adipokine (insulin, leptin, adiponectin, MCP-1, TNF-α and PAI-1), and Cytokine (IL-6 and IFN-γ) kits (Merck Millipore, Billerica, MA, USA), respectively. Homeostatic model assessment of IR (HOMA-IR) and intraperitoneal glucose tolerance test were calculated and performed as described in our previous studies. 17,18
Hepatic and fecal lipid contents
The classical Folch lipid extraction method was used for lipid extraction from liver. 19 The lipid contents of liver and feces were also determined as described in our previous studies. 17,18
Hepatic lipid- and glucose-regulating enzyme activities
Hepatic cytosolic, microsomal, and mitochondrial fractions were prepared according to the Hulcher and Oleson method, 20 and protein levels were determined using the Bradford method. Fatty acid synthase (FAS), 21 phosphatidate phosphohydrolase, 22 carnitine palmitoyltransferase (CPT), 23 and β-oxidation, 24 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), 25 and acyl-CoA:cholesterol acyltransferase (ACAT) 26 activities were measured as previously described. Glucose-6-phosphatase (G6Pase), 27 glucokinase (GK), 28 phosphoenolpyruvate carboxykinase (PEPCK) 29 activities, and glycogen 30 concentrations were also determined as previously described.
Hepatic antioxidant-regulating enzyme activities
The hepatic H2O2 31 and thiobarbituric acid reactive substance (TBARS) 32 contains and plasma and hepatic paraoxonase (PON) 33 activities were measured as previously described.
Morphology of liver, fat, and pancreatic tissues
Liver and epididymal WAT were fixed in paraformaldehyde/phosphate-buffered saline (PBS) (10% v/v); paraffin-embedded sections were stained using hematoxylin and eosin, and Masson's trichrome. The pancreatic tissue samples were fixed in paraformaldehyde/PBS (4% [v/v]; paraffin-embedded sections were immunochemically stained for insulin. The stained areas were analyzed under an optical microscope (Nikon, Tokyo, Japan) at 200 × magnification.
RNA isolation and microarray analysis
Liver, epididymal WAT, and gastrocnemius muscle samples were prepared and total RNA extracted. Microarray analysis was analyzed in epididymal WAT and gastrocnemius muscle and then the differentially expressed gene analysis between groups was performed as described previously. 17
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways mapper was consulted for the analysis of gene functions involved in the tricarboxylic acid (TCA) cycle.
Gene expression analysis
Total RNA of all samples was reverse transcribed to cDNA using a QuantiTect® reverse transcription kit (#205314; Qiagen Gmbh, Hilden, Germany). Real-time RT-PCR was performed using the SYBR Green PCR kit (#204143; Qiagen Gmbh) using the CFX96™ real-time system (Bio-Rad, Hercules, CA, USA). The expression level of specific mouse-GAPDH was set as internal controls. The sequences of all primers are listed in Table 1.
Primer Sequences Used for the Reverse Transcription Quantitative PCR
ACC, acetyl-CoA carboxylase; ADRB3, β-3 adrenergic receptor; CIDEA, cell death-inducing DFFA-like effector A; COL1, collagen 1; COX8β, cytochrome c oxidase subunit 8β; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IRS2, insulin receptor substrate 2; MMP2, matrix metalloproteinase 2; PGC1α, PPARγ coactivator 1α; PPARγ, peroxisome proliferator-activated receptor γ; SCD1, stearoyl-CoA desaturase 1; SIRT1, sirtuin 1; SREBP1α, sterol regulatory element-binding transcription factor 1α; TFAM, transcription factor A, mitochondrial; TIMP1, tissue inhibitors of metalloproteinase 1; TLR4, toll-like receptor 4; UCP1, uncoupling protein 1.
Statistical analysis
Values are represented as means ± standard error of the mean. Statistical analyses were performed using SPSS software (SPSS, Inc., Chicago, IL, USA). Significance was determined by using the Student's t-test.
Results
Fisetin supplementation prevented adiposity and dyslipidemia
FI-supplement markedly suppressed BW and food efficiency ratio despite the increase in food intake compared with those in the HFD-fed mice (Fig. 1A–C). Moreover, the weights of the epididymal, perirenal, retroperitoneum, visceral and total WAT, excluding interscapular WAT, and the epididymal adipose size in HFD+FI group were significantly decreased (Fig. 1D, E). FI significantly decreased the expression of adipocyte genes involved in lipogenesis (e.g., PPARγ, SREBP1, FAS, and SCD1), whereas it upregulated FA oxidation-related gene (e.g., CPT1, CPT2, and COX8β) expressions (Fig. 1F).
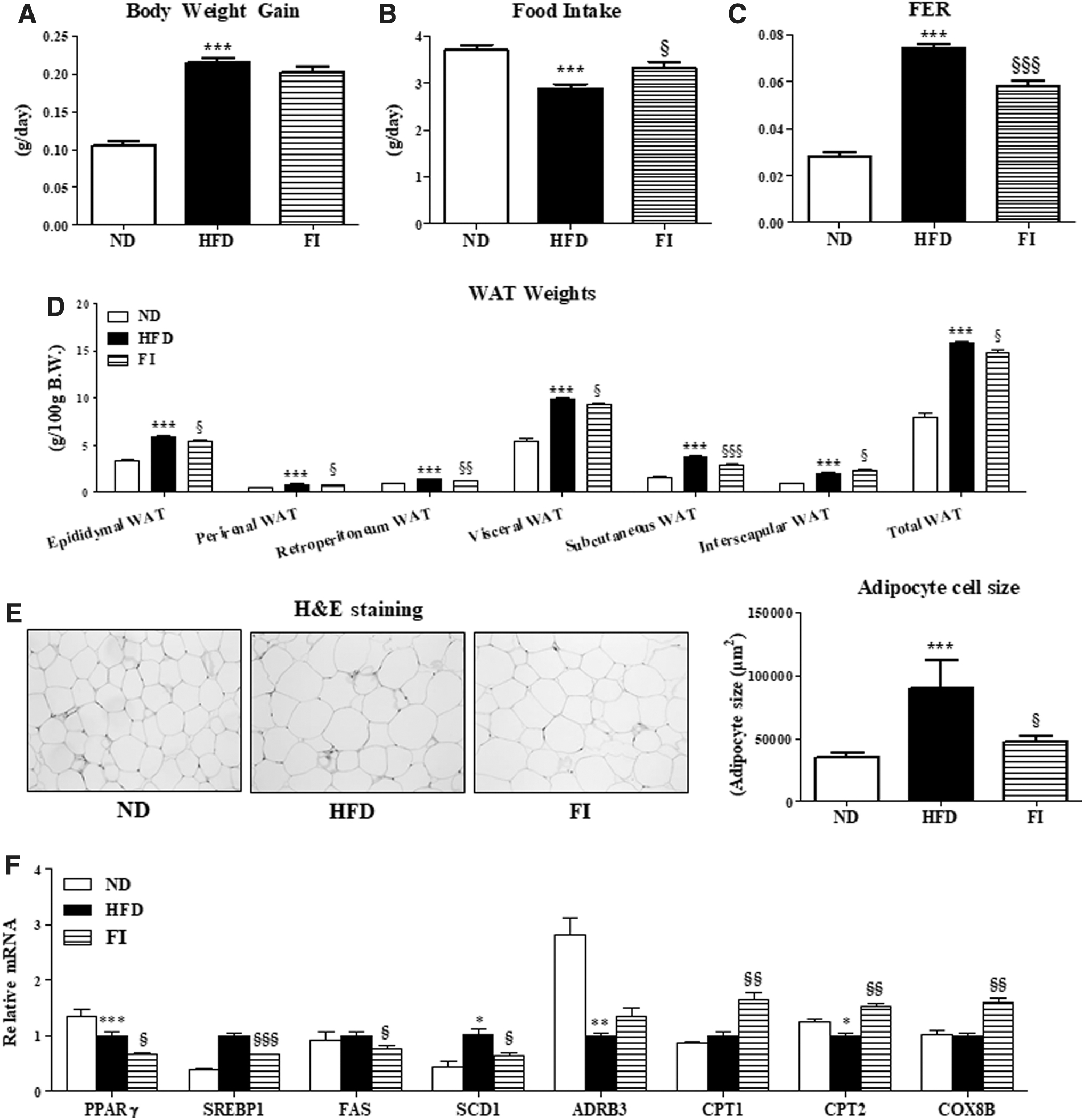
Effect of fisetin supplementation for 16 weeks on body weight
FI-supplemented mice also significantly decreased plasma levels of TC, AI, ApoB, and ApoB/ApoA ratio, while it markedly increased HTR and ApoA levels compared to HFD group. In addition, FI-supplement significantly decreased plasma PLTP activity than HFD group (Fig. 2).

Effect of fisetin supplementation for 16 weeks on plasma lipid levels in HFD-fed mice. The data are presented as means ± SEM. ND (AIN-76) versus HFD; *P < .05, **P < .01, ***P < .001. HFD versus FI (HFD +0.02% FI); § P < .05, §§ P < .01, §§§ P < .001. AI, atherogenic index; Apo, apolipoprotein; FFA, free fatty acid; HDL-C, high-density lipoprotein cholesterol; HTR, HDL-C-to-TC ratio; PLTP, phospholipid transfer protein; TC, total-cholesterol; TG, triglyceride.
Fisetin supplementation increased weights of skeletal muscle and BAT, and upregulated the expression of muscle genes involved in the TCA cycle
The mice fed with FI exhibited significant increases in the weight of BAT and skeletal muscle (Fig. 3A, B). In BAT, FI supplementation enhanced thermogenesis-related genes (Ucp1, Ppargc1a, Sirt1, Cox8β, Cidea, and Tfam) (Fig. 3C). Moreover, FI increased TCA cycle-associated gene expressions, including Dld, Cs, Idh1, Idh2, Suclg1, and Sdhb that were downregulated by HFD (Fig. 3D).

Effect of fisetin supplementation for 16 weeks on the weight of BAT
Fisetin supplementation prevented hepatic steatosis and fibrosis
FI-supplemented mice exhibited a significantly decreased size and number of hepatic lipid droplets, and lower hepatic FA and TG concentrations, as well as liver weights, relative to mice in the HFD group (Fig. 4A–C). Moreover, FI supplementation significantly decreased the activity of enzymes involved in cholesterol synthesis (e.g., HMGCR and ACAT) and lipogenesis (e.g., FA synthase) and gene (e.g., PPARγ, SREBP, and SCD1) expressions, whereas it markedly increased FA oxidation-related enzyme (e.g., CPT and β-oxidation) activities and gene (e.g., CPT, PGC1α, and PGC1β) expressions, in the liver of HFD-fed mice (Fig. 4D, E).

Effect of fisetin supplementation for 16 weeks on liver weights
Furthermore, compared with the HFD group, the levels of hepatic lipotoxicity markers, GOT and GPT, and mitochondrial H2O2 and TBARS were remarkably lower in the FI group (Fig. 5A, B). The activities of plasma and hepatic PON, a hydrolytic enzyme with capability to protect against lipid oxidation, were strikingly increased by FI supplement in HFD-fed mice (Fig. 5C). FI greatly reduced plasma levels of proinflammatory cytokines, including MCP-1, TNF-α, PAI-1, IL-6, and IFN-γ, compared to the HFD group (Fig. 5D). Moreover, FI supplementation not only significantly reduced the hepatic expression of ECM remodeling-associated genes (e.g., COL1, TIMP1, MMP2, MMP3, and MMP9), along with a decrease in mRNA expression of hepatic proinflammatory genes, TLR4 and IL-6 (Fig. 5E). In addition, Masson's trichrome staining of liver and epididymal WAT sections revealed greater collagen accumulation (blue) in HFD mice, but FI showed normal architecture in the liver and very thin and tightly filled collagen sheets surrounding adipocyte (Fig. 5F). In addition to liver, FI resulted in downregulation of the adipocyte genes responsible for ECM remodeling and regulation (Col1a, Mmp3, Mmp23, Tgfbi, Tgfbr1, Tlr4, Ctsa, and Ctgf) and chemokine and cytokine (Tnfrsf12a, Ccl2, Ccl11, Cxcl4, Cxcl14, Cxcr4, and Emr1) in comparison to the HFD group (Fig. 6A, B).

Effect of fisetin supplementation for 16 weeks on plasma GOT and GPT
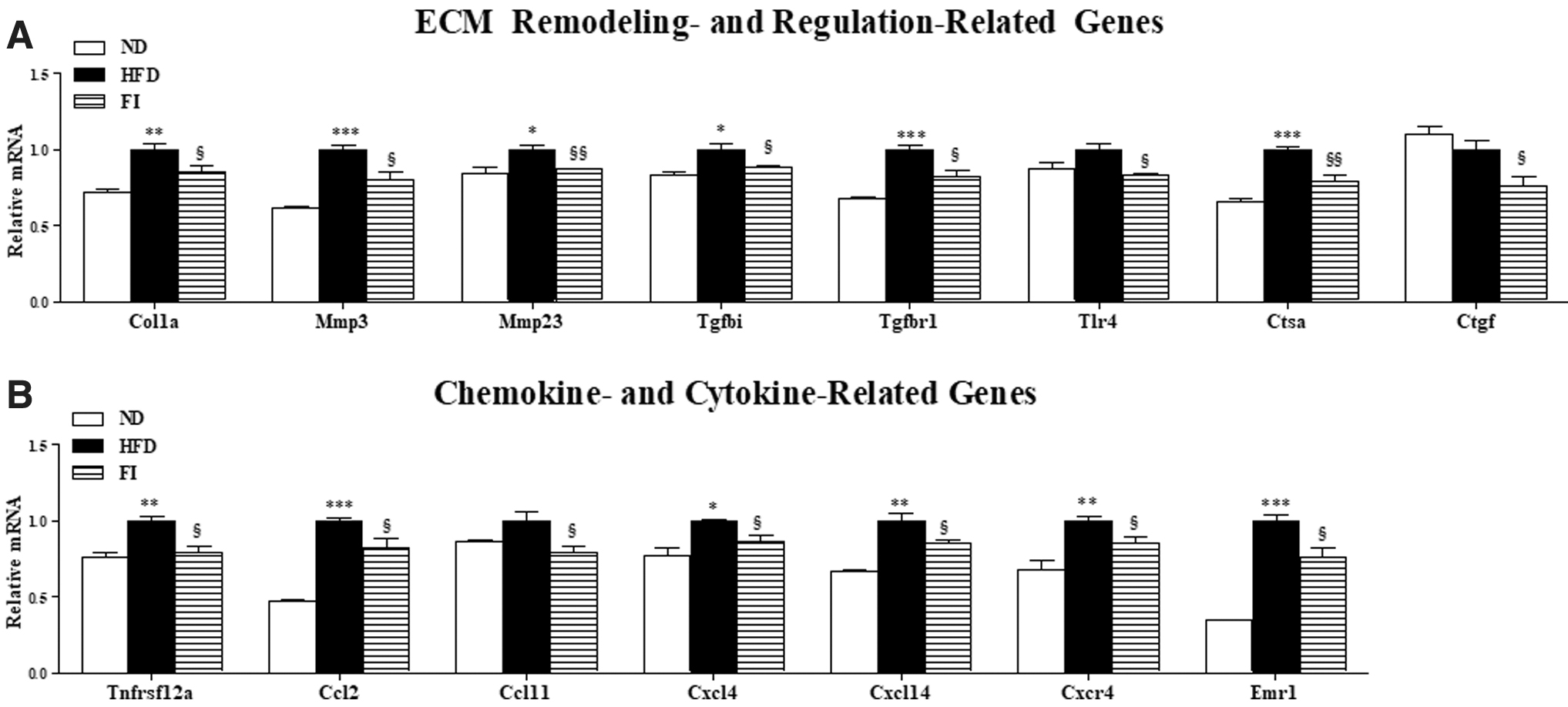
Effect of fisetin supplementation for 16 weeks on the adipocyte gene transcription patterns related to ECM remodeling and ECM regulation
Fisetin supplementation prevented IR, adipokine dysregulation, and pancreatic islet hypertrophy
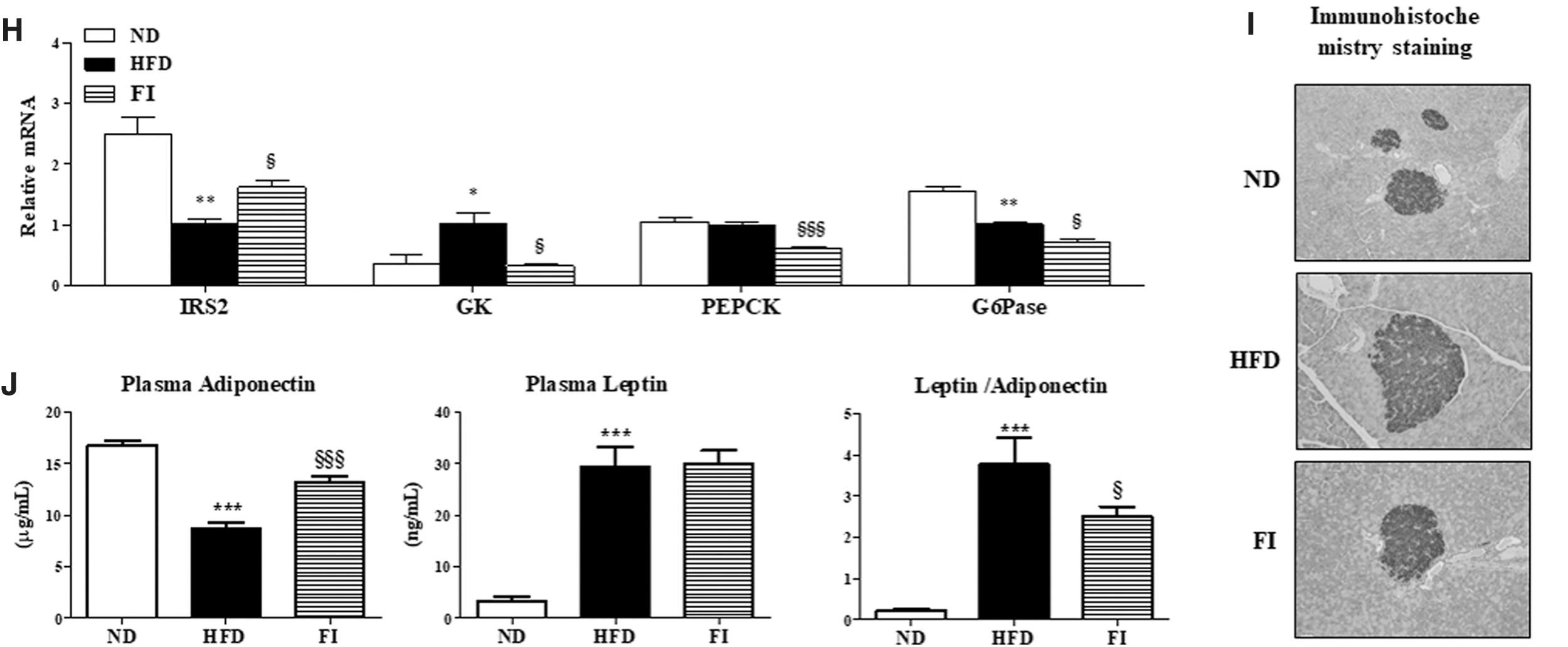
FI markedly decreased the plasma glucose and insulin (Fig. 7A, B) concentrations, and HOMA-IR index and glucose tolerance (Fig. 7C, D) relative to the HFD group. In addition, FI significantly increased the circulating GLP-1 level (Fig. 7E). Glycogen concentrations and GK and PEPCK activities were lowered, while G6Pase/GK ratio was increased by FI compared to mice fed the HFD (Fig. 7F, G). Hepatic mRNA expression also showed that FI supplementation significantly downregulated GK, PEPCK, and G6Pase genes, while it upregulated IRS2 gene in HFD-fed mice (Fig. 7H). Immunohistochemistry staining of the pancreatic tissue for insulin revealed that HFD-induced pancreatic islet hypertrophy was protected by FI supplementation, thereby normalizing the insulin concentration in the plasma (Fig. 7I). Moreover, FI-supplemented HFD-fed mice exhibited greatly increased level of plasma adiponectin, and thus markedly reduced a leptin/adiponectin ratio relative to the mice in the HFD group (Fig. 7J).

Effect of fisetin supplementation for 16 weeks on plasma glucose
Discussion and Conclusions
Being overweight and obese do not only increase adiposity and hepatic steatosis, but in these states the liver tissue accumulate excessive ECM protein, including collagen, causing nonalcoholic steatohepatitis (NASH), a severe form of NAFLD. 34 –37 To date, no effective drugs have been developed to treat patients with NASH and thus, in the hepatic steatosis stage, the understanding of the molecular mechanisms that cause oxidative balance impairment, lipid accumulation, fibrosis in the liver, and IR would create an opportunity to prevent and attenuate the development of NAFLD and/or NASH.
Adiposity is linked to the increased production and release of several proinflammatory cytokines and ECM components, which contribute to chronic low-grade inflammation and obesity-linked metabolic disorders such as IR and fibrosis. 18,38,39 In the present study, FI ameliorated adiposity, as evidenced by a decrease in WAT weight, by increasing expression of genes involved in FA oxidation (e.g., CPT1, CPT2, and COX8β), while decreasing the expression of genes involved in lipogenesis in adipose tissue (e.g., PPARγ, SREBP1, FAS, and SCD1). Moreover, we observed that FI supplementation significantly increased BAT ad skeletal muscle weights. FI enhanced the expression of not only thermogenesis-related genes (e.g., Ucp1, Ppargc1a, Sirt1, Cox8β, Cidea, and Tfam) in BAT, but also TCA cycle-associated genes (e.g., Dld, Cs, Idh1, Idh2, Suclg1, and Sdhb) in skeletal muscle, which may be related to the decreased adiposity and NAFLD in the HFD-fed obese mice. BAT and skeletal muscle are major organs that can generate heat by burning fat, which can be a potential therapeutic target for obesity and its related metabolic diseases, including NAFLD. 6 –8,40 –44
The improved adiposity by FI was likely associated with a reduction of plasma proinflammatory cytokine levels (e.g., MCP-1, TNF-α, PAI-1, IL-6) and gene expressions involved in ECM remodeling and inflammation in adipose tissue as well as liver, leading to the prevention of fibrosis. Hepatic fibrosis is a common wound healing reaction to chronic liver damage, 45 involving extra deposition ECM proteins. 46,47 The fibrotic ECM comprised collagens, mainly type I and type III collagens, 46 and overexpression of tissue inhibitors of metalloproteinases (TIMPs) and matrix metalloproteinases (MMPs), which are the main regulators of ECM turnover in hepatic fibrosis, contribute to ECM deposition and fibrosis progress. 48,49 In addition, FI increased the activities of PON in plasma and liver, resulting in a decrease in excessive ROS production as evidenced by lower hepatic TBARS and mitochondrial H2O2 levels, which reflects improved hepatic fibrosis and IR.
The antifibrotic effects coupled with the decreased adiposity by FI supplementation were strongly linked to the improvement of NAFLD and IR. The FI group exhibited a significant decrease in hepatic lipid levels by increasing FA oxidation, and decreasing lipogenesis compared to the HFD group, which in turn caused a significant decrease in the hepatic fibrosis, which may contribute to the prevention of IR. FI prevented pancreatic islet hypertrophy, and which is known to contribute to IR by increasing insulin secretion per β cell to compensate for insulin desensitization. 50 Furthermore, relative to the HFD mice, FI supplementation markedly increased the plasma level of GLP-1, which can exert anti-inflammatory effects. 51 Dysregulated hepatic glucose metabolism caused by an increase in gluconeogenesis also reported to contribute to the development of IR. 52 Thus, the suppression of gluconeogenesis by the decreased expression and lower enzymatic activity of hepatic PEPCK appears to be associated with the enhanced insulin sensitivity observed in FI-supplemented obese mice. Moreover, FI supplementation significantly increased G6Pase/GK ratio. The G6Pase/GK ratio roughly reflects the balance between hepatic glucose release and uptake, and it is remarkably reduced in obese type II diabetics. 53 In addition, we determined the plasma PLTP activity to investigate the antihyperlipidemic effect of FI supplementation. Our results indicate that FI supplementation increased the plasma PLTP activity with a simultaneous decrease in the plasma TC and AI levels. Several studies have demonstrated that plasma PLTP activity is positively associated with IR and PLTP might be a marker for NAFLD. 54,55 Therefore, our results suggest that FI may have a protective role in dyslipidemia, which contributes to the development of IR and NAFLD.
In conclusion, our observations using diet-induced obese mice demonstrate a novel mechanism of FI action in improving obesity and related metabolic disorders such as hyperlipidemia, hepatic steatosis, fibrosis, and IR. Dietary FI improved hepatic steatosis by restricting lipogenesis, while promoting lipolysis in the liver. FI also prevented adiposity via an increase in the expression of genes involved in FA oxidation and the decrease in the expression of genes involved in lipogenesis in WAT, which related to the prevented NAFLD. Moreover, FI enhanced mRNA expression of thermogenesis- and TCA-cycle-related genes in BAT and skeletal muscle, respectively, along with increased BAT and skeletal muscle weights, which may lead to reduced adiposity and NAFLD in HFD-induced obese mice. This is the first study investigating the effect of FI on BAT and muscle in relationship to obesity and its metabolic diseases such as NAFLD/NASH. FI supplementation also decreased excessive ROS production by increasing PON activity, adipokine dysregulation, proinflammatory cytokine production, and ECM accumulation in the liver, which contributed to improvement of hepatic lipotoxicity and fibrosis. FI supplementation improved IR, in part, by normalizing pancreatic islet dysfunction, and it decreased hepatic gluconeogenesis and proinflammatory responses. Taken together, the present findings indicate that FI may be useful for ameliorating the deleterious effects of HFD-induced obesity and related metabolic complications such as hyperlipidemia, NAFLD, hepatic fibrosis, and IR.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the BK21 Plus Program (Department of Food Science and Nutrition, Kyungpook National University, 22A20130000161), and a National Research Foundation (NRF) of Korea grant funded by the Korea government (MSIP) (NRF-2020R1I1A3074694 and NRF-2019R1C1C1009530).