
Abstract
Idiopathic pulmonary fibrosis (IPF) is a lung disease that results in scarring of the lungs for an unknown reason. Although many studies have been conducted on IPF, precise mechanisms and treatments have not yet been identified. In this study, we found that aucuparin, a natural product isolated from Sorbus aucuparia, inhibited pulmonary fibrosis in a bleomycin (BLM)-induced lung fibrosis mouse model. In the lung samples of mice treated with aucuparin, the gene expression of inflammation and macrophage activation markers was reduced compared to those treated with BLM alone. Moreover, aucuparin decreased the expression of profibrotic marker genes and increased the expression of antifibrotic marker genes. Finally, we observed that aucuparin significantly suppressed transforming growth factor-β-induced activation of inflammatory cytokine production and collagen synthesis from macrophages and fibroblasts, respectively. Taken together, these data demonstrate that aucuparin inhibits lung fibrosis via its anti-inflammatory action and support its potential to be a therapeutic drug for IPF treatment.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a special form of chronic interstitial pneumonia of unknown origin. 1 It occurs mainly in the elderly and is usually associated with the histopathological form of usual interstitial pneumonia. 2 In individuals with genetic factors, or in the elderly, long-term exposure to smoking, dust, gastroesophageal reflux, or other environmental factors causes abnormal pathways to be activated, preventing normal wound healing. 3 IPF usually has a poor prognosis, with an average life expectancy of about 3 years after diagnosis, and most of the causes of death are due to the disease itself, such as progression of respiratory failure. 4 Although pirfenidone and nintedanib are used as therapeutic drugs to treat IPF, they are still insufficient to cure patients. 5,6
Although the mechanism of IPF has not been fully elucidated, many studies have shown that macrophages and fibroblasts activated by external stimuli may affect IPF pathogenesis. 7 First, activated macrophages induce differentiation and proliferation of fibroblasts by secreting chemokines. 8,9 Activated fibroblasts produce collagen and matrix metalloproteinases (MMPs), which cause excessive accumulation in lung tissue and disrupt tissue structure. 3,10 Subsequently, activated macrophages and fibroblasts affect each other, causing the lung to exhibit irreversible pathogenic phenotypes. 11
Aucuparin is a biphenyl phytoalexin that was first isolated from the heartwood tree of Sorbus aucuparia. 12,13 Aucuparin has been reported to have anti-inflammatory activity. 12,13 It suppressed the production of the N-formyl-methyl-leucyl-phenylalanine (fMLP)-induced generation of the superoxide anion, an inflammatory mediator produced by neutrophils. 14 However, there has been no study on whether aucuparin is effective in treating pulmonary fibrosis.
In this study, we observed that aucuparin inhibited pulmonary fibrosis using a bleomycin (BLM)-induced lung fibrosis mouse model. In the mouse samples treated with aucuparin, the expression of inflammatory genes and activated macrophage marker genes was decreased. We also confirmed that the expression of collagen and MMPs was significantly reduced in aucuparin-treated mouse lung tissues. Finally, we showed that aucuparin significantly suppressed transforming growth factor (TGF)-β-induced activation of inflammatory cytokine production and collagen synthesis from macrophages and fibroblasts, respectively. These data suggest that aucuparin inhibits pulmonary fibrosis by suppressing the activation of macrophages and fibroblasts, and may be a potential therapeutic agent for IPF treatment.
Materials and Methods
Animal studies
The study protocol for animal experiments was approved by the Institutional Animal Care and Use Committee of Yonsei University College of Medicine (Certification No. IACUC-2018-0087). Eight-week-old C57BL/6 male mice were housed under pathogen-free conditions at room temperature using a 12-h light-dark cycle.
For the BLM-induced lung fibrosis mouse model, mice were intratracheally injected with [Phosphate buffer saline (PBS), control] or 2 mg/kg BLM (Santa Cruz Biotechnology, Santa Cruz, CA). Five to nine mice were used for each group. After treatment with BLM, aucuparin was administered intraperitoneally every other day for 2 weeks with a low dose of 8 or 12 mg/kg. After injecting aucuparin for 2 weeks, the mice were sacrificed. Bronchoalveolar lavage fluid and lungs of mice were harvested and analyzed.
RNA isolation and quantitative reverse transcription-polymerase chain reaction
Total RNA from mouse lung tissues and cells was prepared using Ribospin II (GeneAll, Korea). RNA samples were reverse-transcribed using CellScript (CellSafe, Yongin, Korea) following the manufacturer's protocol. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed using SYBR Green PCR Master Mix reagents and an ABI Prism 7700 Sequence Detection System (Applied Biosystems, Carlsbad, CA). The following primer sequences were used for qRT-PCR: Cox-2 5′-CAGACAACATAAACTGCGCCTT-3′ and 5′-GATACACCTCTCCACCAATGACC-3′, iNOS 5′-CGAAACGCTTCACTTCCAA-3′ and 5′-TGAGCCTATATTGCTGTGGCT-3′, IL-1β 5′-CCTTCCAGGATGAGGACATGA-3′ and 5′-TGAGTCACAGAGGATGGGCTC-3′, Arg1 5′-AACACGGCAGTGGCTTTAACC-3′ and 5′-GGTTTTCATGTGGCGCATTC′-3′, Cd206 5′-GTTCACCTGGAGTGATGGTTCTC-3′ and 5′-AGGACATGCCAGGGTCACCTTT-3′, Cd163 5′-GGCTAGACGAAGTCATCTGCAC-3′ and 5′-CTTCGTTGGTCAGCCTCAGAGA-3′, Acta2 5′-ACTGGGACGACATGGAAAAG-3′ and 5′-GTTCAGTGGTGCCTCTGTCA-3′, Pai-1 5′-CCTTGCTTGCCTCATCCTGG-3′ and 5′-CTGGAAGAGCTTGAAGAAGTGG-3′, Col1a1 5′-CCTCAGGGTATTGCTGGACAAC-3′ and 5′-CAGAAGGACCTTGTTTGCCAGG-3′, Col3a1 5′-GACCAAAAGGTGATGCTGGACAG-3′ and 5′-CAAGACCTCGTGCTCCAGTTAG-3′, Mmp9 5′-GTCTTCCTGGGCAAGCAGTA-3′ and 5′-CTGGACAGAAACCCCACTTC-3′, Timp3 5′-AGCCAAAGCAGTGAGCGAGAAG-3′ and 5′-GCCGTGTAGATAAACTCGATGTC-3′, Cxcl10 5′-ATCATCCCTGCGAGCCTATCCT-3′ and 5′-GACCTTTTTTGGCTAAACGCTTTC-3′, Il-6 5′-GAGGATACCACTCCCAACAGACC-3′ and 5′-AAGTGCATCATCGTTGTTCATACA-3′, Tnfa 5′-GGTGCCTATGTCTCAGCCTCTT-3′ and 5′-GCCATAGAACTGATGAGAGGGAG-3′, Gapdh 5′-CCCATCACCATCTTCCAGGAGC-3′ and 5′-CCAGTGAGCTTCCCGTTCAGC-3′. The cDNA concentration was normalized using Gapdh. Each sample was analyzed in triplicate for each target gene.
Western blot analysis
Mouse lung tissues were mixed with stainless steel beads and lysis buffer (20 mM Tris-Cl, 150 mM NaCl, 1% Triton X-100, 1.5% MgCl2, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail, pH 7.5) in 2 mL Safe-Lock microtube. The samples were subjected to bead beating in triplicate using the TissueLyserII (Qiagen, Hilden, Germany) at a frequency of 30 Hz for 3 min each, with a 5 min rest on ice, followed by centrifugation. The supernatant was then transferred to a new microtube. Protein concentrations were quantified using a 660 nm protein assay reagent (Thermo Scientific, Rockford, IL). Equal amounts of protein extracts were subjected to electrophoresis on SDS-polyacrylamide gels and then transferred to nitrocellulose transfer membranes (Whatman, Dassel, Germany). The membranes were blocked in Tris-buffer (pH 7.4) containing 0.1% (v/v) Tween 20 (Sigma-Aldrich) and 0.05 g/ml Difco™ skim milk (BD Biosciences, Sparks, MD) and probed with primary antibodies. The following antibodies were used: COX-2 (Santa Cruz Biotechnology, Dallas, TX), iNOS (Santa Cruz Biotechnology, Dallas, TX), α-SMA (Abcam, Cambridge, UK), PAI-1 (Santa Cruz Biotechnology, Dallas, TX), and β-actin (Santa Cruz Biotechnology, Dallas, TX).
Masson's trichrome staining
The experiment used the same procedures as previously reported. 15 The blue-stained collagen portions were quantified using ImageJ software in a × 200 field image area (Olympus, Tokyo, Japan).
Sircol collagen assay
Collagen contents in mouse lung tissues were analyzed using the Sircol Collagen Assay Kit (Biocolor, Carrickfergus, UK) following the manufacturer's protocol.
Immunocytochemistry
The harvested lungs of mice were fixed and embedded in paraffin and sectioned. Following the rehydration and blocking step (DAKO, Glostrup, Denmark), immunofluorescent staining was performed. Anti-CD206 (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-NF-kB p65 (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies were used as the primary antibodies. The secondary antibody was anti-goat 488 (Abcam). Nuclei were counterstained with DAPI (DAKO). Tissues were imaged and analyzed using a Zeiss LSM700 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Cell culture and reagents
Mouse alveolar macrophage cell line MH-S and mouse fibroblast cell line MLg were purchased from the Korean Cell Line Bank (Seoul, Korea). MH-S cells were cultured in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS), 1% antibiotic/antimycotic solution (Corning, Manassas, VA), and 0.05 mM β-mercaptoethanol at 37°C in 5% CO2. MLg cells were cultured in DMEM supplemented with 10% FBS and 1% antibiotic/antimycotic solution. Aucuparin was purchased from ChemFaces (Wuhan, Hubei, China). TGF-β 1 was purchased from ProSpec (East Brunswick, NJ).
Statistical analyses
Statistical analyses were performed using GraphPad Prism 6.0 (San Diego, CA). One-way analysis of variance was used to compare four independent groups. The significance levels are indicated as follows: not significant, P > .05; *P < .05; **P < .01; and ***P < .001.
Results
Protective effects of aucuparin on BLM-induced lung fibrosis
In previous studies, aucuparin was shown to have an anti-inflammatory effect. 13,14,16 To examine the antifibrotic effects of aucuparin, a BLM-induced lung fibrosis mouse model was used (Fig. 1A). BLM-induced lung fibrosis is the most common experimental model of human lung fibrosis. 17 After BLM injection, the mice were treated with aucuparin every other day for 2 weeks. BLM intratracheal injection resulted in severe destruction of the lung structure and irreversible lung fibrosis compared to the PBS-treated group (Fig. 1B). Treatment with aucuparin significantly inhibited the progression of lung fibrosis and accumulation of collagen. The antifibrotic effect of aucuparin was more effective in the group with high concentrations of aucuparin. Masson's trichrome staining was quantified using ImageJ software (Fig. 1C). Total collagen content of mouse lung was measured by the Sircol collagen assay, and collagen secretion was highly reduced when aucuparin was administered in BLM-treated mice (Fig. 1D). Next, we differentially counted the macrophages, neutrophils, lymphocytes, and eosinophils of the total cells in bronchoalveolar lavage (BAL) fluid. After BLM treatment, the number of neutrophils increased significantly, compared to PBS-treated control mice. Lymphocytes and eosinophils in the BAL fluid of all groups were negligible. BLM treatment caused a significant increase in the total cell count of the BAL fluid as compared to the control; however, it was significantly reduced following aucuparin treatment (Fig. 1E, F). Collectively, these data demonstrate that aucuparin inhibits lung fibrosis by suppressing excessive collagen deposition.
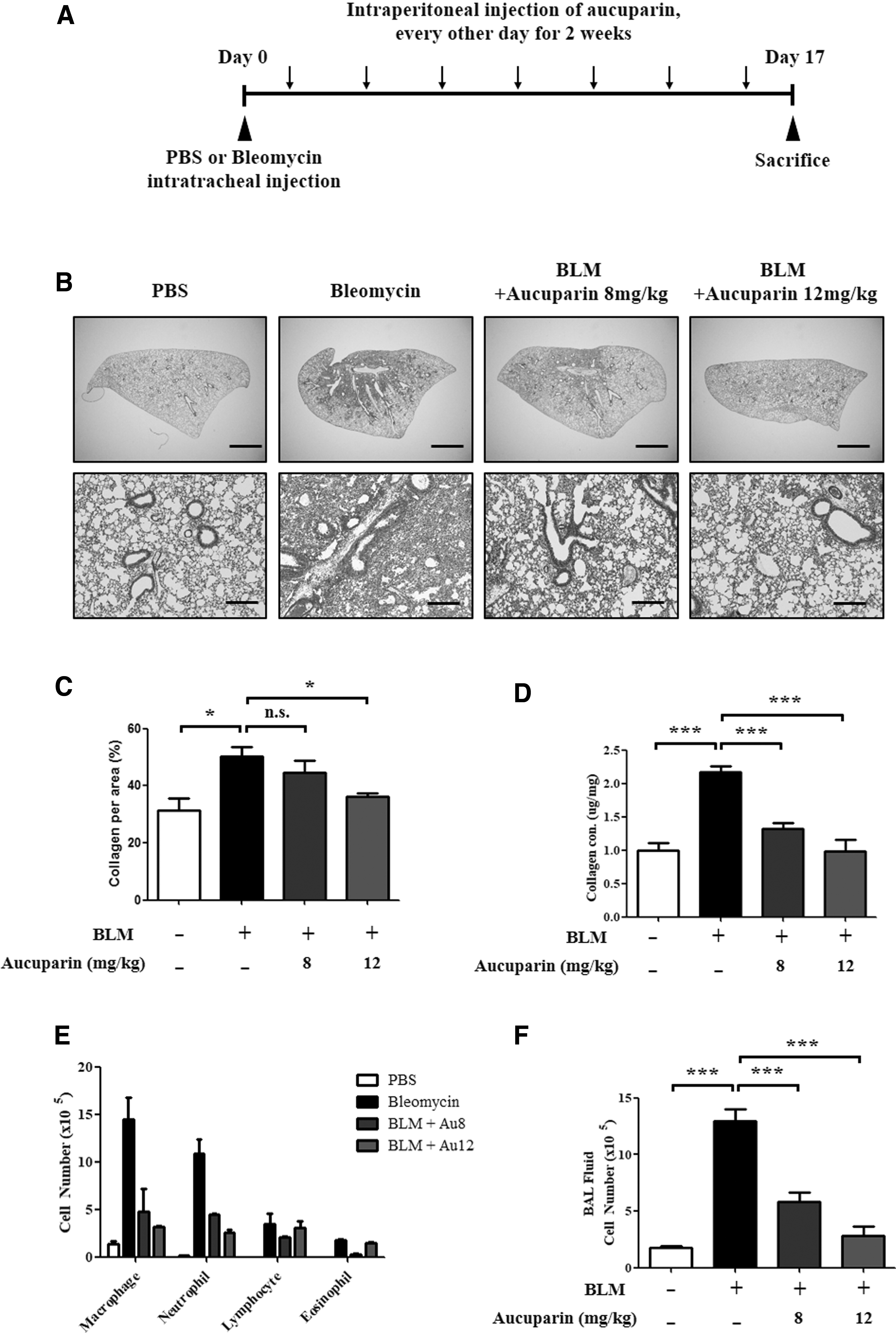
Aucuparin inhibits pulmonary fibrosis significantly in BLM-induced lung fibrosis mouse model.
Aucuparin suppresses the expression of inflammatory genes and activates macrophage markers in lung tissues of BLM-treated mice
Given the data suggesting that aucuparin inhibits lung fibrosis progression, we investigated whether aucuparin regulates inflammatory gene expression in lung tissues of BLM-treated mice. As shown in Figure 2A, the expression levels of COX-2 and iNOS proteins were significantly increased in lung tissues after BLM treatment, whereas administration of aucuparin suppressed the expression of COX-2 and iNOS protein in a dose-dependent manner. The mRNA levels of Cox-2, iNos, and Il-1β were also decreased in the lung tissues of aucuparin-treated mice (Fig. 2B).
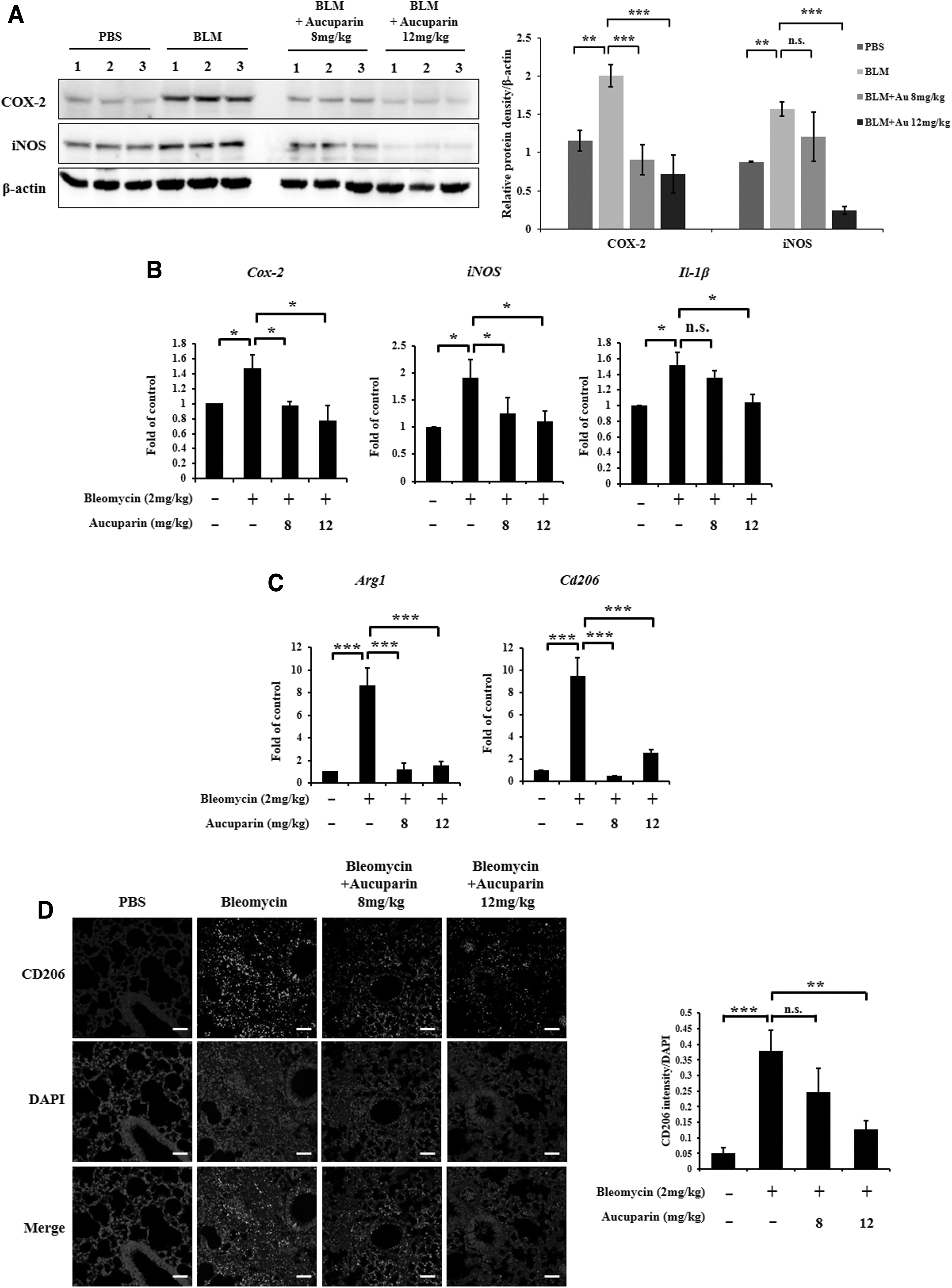
The inhibitory effects of aucuparin on the expression of inflammatory genes.
Next, we investigated whether the anti-inflammatory effects of aucuparin are caused by preventing the activation of macrophages. The mRNA levels of activated macrophage markers Arg1 and Cd206 were suppressed in the lung tissue of aucuparin-treated mice (Fig. 2C). Moreover, immunofluorescence staining for a CD206 antibody confirmed that CD206 expression levels were decreased following the administration of aucuparin compared to the control (Fig. 2D). Quantification of immunofluorescence staining in lung tissues revealed that CD206 protein was increased in BLM-treated mice and downregulated following aucuparin treatment. These data indicate that aucuparin suppresses the expression of inflammatory genes and the activation of profibrotic macrophages.
Aucuparin suppresses activation of myofibroblast and collagen synthesis in mouse fibrotic lung tissues
Activated macrophages cause fibroblasts to differentiate into myofibroblasts and secrete cytokines to induce protein expression of collagens and MMPs. 18,19 We investigated whether aucuparin inhibits the differentiation of fibroblast to myofibroblast. BLM treatment increased protein and mRNA expression levels of the myofibroblast markers α-SMA and fibrotic marker PAI-1; however, it was significantly decreased by aucuparin treatment (Fig. 3A, B).
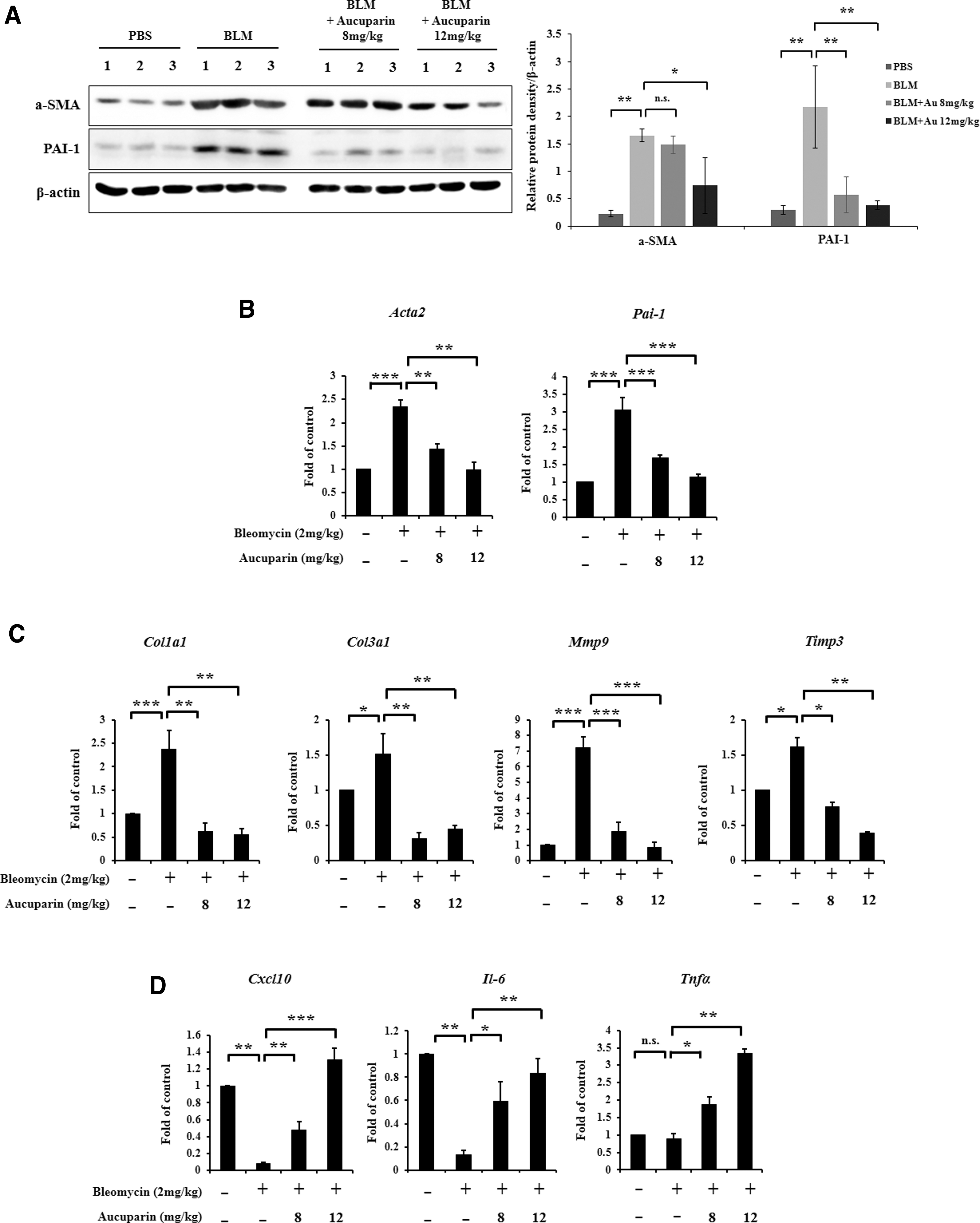
Antifibrotic effects of lung tissue in BLM-induced pulmonary fibrosis by aucuparin.
Next, we examined whether aucuparin inhibits the expression of collagen and MMPs. As shown in Figure 3C, qRT-PCR analysis demonstrated that the expression of profibrotic markers, including Col1a1, Col3a1, Mmp9, and Timp3 were elevated in BLM-induced mice relative to controls. However, the BLM-induced activation of profibrotic markers was completely reversed by aucuparin treatment. Conversely, the expression of antifibrotic markers Cxcl10, Il-6, and Tnfα was elevated following aucuparin treatment, indicating that aucuparin inhibits pulmonary fibrosis by reprogramming profibrotic markers to antifibrotic markers in macrophages (Fig. 3D).
Aucuparin inhibits TGF-β-induced activation of inflammatory cytokine production and collagen synthesis
The aim was to determine whether aucuparin suppresses proinflammatory cytokine production from alveolar macrophages. It is well known that TGF-β is the most potent profibrotic mediator and modulates pulmonary fibrosis through the recruitment and activation of profibrotic macrophages and fibroblasts. 20 Alveolar macrophage MH-S cells were incubated with TGF-β 1, a major profibrotic cytokine, to induce the activation of pro-inflammatory cytokine genes. TGF-β treatment increased the expression of proinflammatory cytokines in MH-S cells; however, aucuparin inhibited TGF-β-induced expression of proinflammatory cytokines in a dose-dependent manner (Fig. 4A). Similar to the results of mouse lung tissue, TGF-β-induced expression of activated macrophage markers, including Arg1 and Cd206, was reversed by aucuparin treatment (Fig. 4A). This indicates that aucuparin inhibits TGF-β-induced proinflammatory cytokine production in macrophages. It is known that NF-κB is activated and enters into the nucleus to regulate the induced transcription of proinflammatory genes, a process known as NF-κB nuclear translocation. 15 Immunofluorescence staining of NF-κB p65 indicated that upon TGF-β treatment, the NF-κB protein translocated to the nucleus (Fig. 4B). However, aucuparin treatment abolished the phenotype induced by TGF-β, resulting in decreased expression of NF-κB in the nucleus.

Effects of aucuparin on activation of macrophages and fibroblasts in response to TGF-β.
We next investigated whether aucuparin inhibits the activation of myofibroblasts and collagen synthesis. In lung fibroblast MLg cells, aucuparin inhibited the TGF-β-induced induction of Acta2 and Pai-1 expression (Fig. 4C). In addition, the increased expression of collagen1/3, Mmp9, and Timp3 by TGF-β-treatment were dosage-dependently inhibited by aucuparin. These data indicate that aucuparin suppresses pulmonary fibrosis by inhibiting proinflammatory cytokine production from profibrotic macrophages.
Discussion
In recent years, many studies have been designed to explore protective and therapeutic bioactive compounds for targeting inflammation and oxidative stress for pulmonary fibrosis. 21 The beneficial activities of phytochemicals have been attributed to the inhibition of leukocyte infiltration, oxidative stress, and proinflammatory cytokine levels, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, as well as the reduction of hydroxyproline content, which is considered an important indicator to directly reflect collagen content in pulmonary fibrosis. 22 Among various phytochemicals, biphenyl compound families such as aucuparin are known to have antimicrobial and antioxidative effects, 12,13 but the efficacy and mechanism of lung fibrosis have not been studied. Based on this, we investigated the antifibrotic effects of aucuparin in vivo lung fibrosis model to assess the therapeutic potential of this compound in pulmonary fibrosis.
IPF is characterized by fibroblast proliferation, excessive collagen accumulation, and other deposition of extracellular matrix (ECM) proteins within the pulmonary interstices resulting in a loss of pulmonary function, and eventually, respiratory failure. 1 Although the etiology of IPF has not been clarified yet, inflammation, oxidative stress, and damage from cytokines are involved in the pathogenesis mechanism. 23 Excessive deposition of ECM proteins, presence of fibroblast foci, and areas of fibrosis next to areas with normal lung parenchyma, or the so-called spatially heterogeneous fibrosis, are the histological hallmarks of IPF. 3,23 Fibrosis is the end-result of exaggerated wound repair and tissue remodeling, which is believed to be caused by cyclical epithelial injury leading to chronic inflammation, and finally fibrosis. Both innate and adaptive immune systems in the body are involved in the development of fibrosis. 24
Among several inflammatory cellular components, macrophages play a crucial role and provide various soluble factors that promote the onset and progression of fibrosis by inducing multiple biological processes, including epithelial mesenchymal transition. 7 Pulmonary macrophages are crucial sentinels that are integral to pulmonary host defense. 25 Macrophages are highly plastic cells, and changes in environmental cues and molecular mediators can shift macrophages from M1 to M2 phenotype. 26 M1 macrophages produce proinflammatory cytokines such as TNF-α, IL-1, and IL-6, thereby maintaining tissue inflammation. 27 M2 macrophages harbor anti-inflammatory and profibrotic properties. 29 In chronic inflammatory conditions, proinflammatory M1 macrophages slowly change into a more anti-inflammatory (profibrotic) M2 phenotype, secreting mediators that promote wound healing. 30 As expected, M2 macrophages are widely present in fibrotic lungs. 7,26 M2 macrophages secrete several growth factors, including TGF-β, fibroblast growth factor, platelet-derived growth factor-α, insulin-like growth factor 1, and vascular endothelial growth factor. M2 macrophages also contribute to the formation of the ECM, specifically to promote collagen synthesis. 26 –28 Therefore, the regulation of macrophage activation can be regarded as an important factor that can control the progression of pulmonary fibrosis; the hypothesis was confirmed by recent studies on phytochemical macrophage regulation. 9,22
The results of our study also confirmed the increase in markers of M2 macrophages (Arg1, CD206) in the lung tissues of mice by BLM; however, this induction was significantly prevented by aucuparin treatment. Conversely, the reduced expression of antifibrotic markers (Cxcl10, Il-6, and TNF-α) by BLM treatment was completely reversed by aucuparin treatment. Moreover, aucuparin efficiently inhibited the TGF-β-induced activation of M2 marker genes as well as proinflammatory cytokine genes in macrophages. This suggests that the inhibitory effect of aucuparin on pulmonary fibrosis may reprogram M2-like fibrosis-inducing macrophages into fibrosis-suppressing macrophages, resulting in significant reduction of profibrotic cytokines and collagen synthesis.
In summary, this study demonstrated the protective role of aucuparin in lung fibrosis models. Aucuparin suppressed fibrosis and the production of proinflammatory cytokines in the lung tissue of the pulmonary fibrosis model induced by BLM. In addition, we showed that aucuparin suppresses the production of proinflammatory cytokines from TGF-β-activated macrophages, and collagen synthesis from fibroblasts. These results suggest that aucuparin may have prophylactic or therapeutic potential in pulmonary lung fibrosis.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the National Research Foundation of Korea (NRF) MRC grant funded by the Korean government (MSIT) (No. NRF-2018R1A5A2025079 and 2020R1A2C3003303 to H.G.Y)