
Abstract
Ginger (Zingiber officinale) is one of the most commonly consumed botanical foods. Owing to its anti-inflammatory and antiviral properties, ginger has been widely used as a homemade remedy during the corona virus disease 2019 (COVID-19) pandemic; however, the mechanisms of its therapeutic activities against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) remain obscure. In this study, we used a drug-likeness approach to screen the active compounds of ginger. Next, we identified candidate targets of active compounds responsible for the anti-SARS-CoV-2 effects of ginger using chemical similarity searching and SARS-CoV-2-human protein–protein interaction (PPI) data. Finally, we analyzed PPIs, Gene Ontology enrichment, and Kyoto Encyclopedia of Genes and Genomes pathway enrichment of the candidate proteins using different bioinformatics tools. A network comprising ginger compounds, human proteins, and SARS-CoV-2 proteins was built through Cytoscape 3.3. The results indicate that the anti-SARS-CoV-2 activity of ginger involves 20 active compounds, 18 potential human targets, and 12 SARS-CoV-2 proteins. These form a pharmacological network in which sigma nonopioid intracellular receptor 1 (SIGMAR1) and histone deacetylase 2 may be druggable hub proteins. In addition, molecular docking showed that 8-gingerdione and dihydrocapsaicin may preferentially interact with SIGMAR1, which was confirmed by further molecular dynamics simulation (150 ns) experiments. In conclusion, ginger targets multiple human proteins and affects multiple SARS-CoV-2 proteins to exert anti-COVID-19 effects. Although further experimental verification is needed, this study provides a quick visual overview of the anti-SARS-CoV-2 action of ginger.
Introduction
The corona virus disease 2019 (COVID-19) pandemic, which is due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has been raging globally for 3 years and remains a social and public health disaster. 1,2 There have been more than 664 million cases and 6.7 million deaths. The wave of Omicron, one of the new SARS-CoV-2 variants, is characteristic with a very high infection rate and a much higher proportion of asymptomatic infections. 3 –5 Due to the failure to eliminate SARS-CoV-2 and its high likelihood of coexisting with humans for a long time, COVID-19 will persist and likely become a recurrent seasonal disease after the Omicron wave. 5 The pandemic remains unpredictable and is moving toward a far more complicated new phase; thus, health systems and societies will have to adapt. 1,2,5,6
Home remedies have the advantages of favorable effects, low cost, and wide availability. Home remedies for COVID-19, including family plant-based and immune-related foods, present a primary health care strategy. 7,8 Ginger (Zingiber officinale), one of the most commonly consumed natural spices, is a good example. Owing to its analgesic, antinausea and antivomiting, antiviral, and anti-inflammatory actions, ginger has been widely used as a home-based medicine to treat influenza and other diseases around the world since ancient times. 9
Ginger has been widely used in the COVID-19 pandemic. Consumption of ginger and garlic in daily life has remarkably increased in an effort to treat influenza and other respiratory symptoms in many areas during the COVID-19 pandemic. 7 Consumption of ginger increased by 33% and peaked during the first and second COVID-19 waves. 10 Ginger is reportedly one of the most frequently used plant-based home remedies administered to COVID-19 patients in Bangladeshi, 11 Latin America and the Caribbean, 12 the Balkan Peninsula, 13 and Ghana and South Africa. 14
Astragalus, honeysuckle, and ginger have been applied to clinical practice for COVID-19 treatment. 15 Licorice, ginger, and peony are traditional medicines that have been mostly used to treat plague in the book titled Wenyilun and Guangwenyilun. 16 Gingerol is known to bind well to the spike protein of SARS-CoV-2 and block viral entry. 17 However, the identification of the anti-SARS-CoV-2 targets of ginger is needed.
In the current work, the active components of ginger were screened using the TCMSP database, and potential targets were fished using chemical similarity measures. SARS-CoV-2–human protein interactions were mined from the PubMed database. Protein–protein interactions (PPIs), Gene Ontology (GO) enrichment, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of the potential targets of ginger that overlap with human proteins that physically interact with SARS-CoV-2 were analyzed using different bioinformatics tools. A drug–target network was established to offer a systematic overview of the potential pharmacological activities of ginger against SARS-CoV-2. Finally, the candidate targets of interest were checked through using molecular docking analyses. A flow process diagram is illustrated in Figure 1.
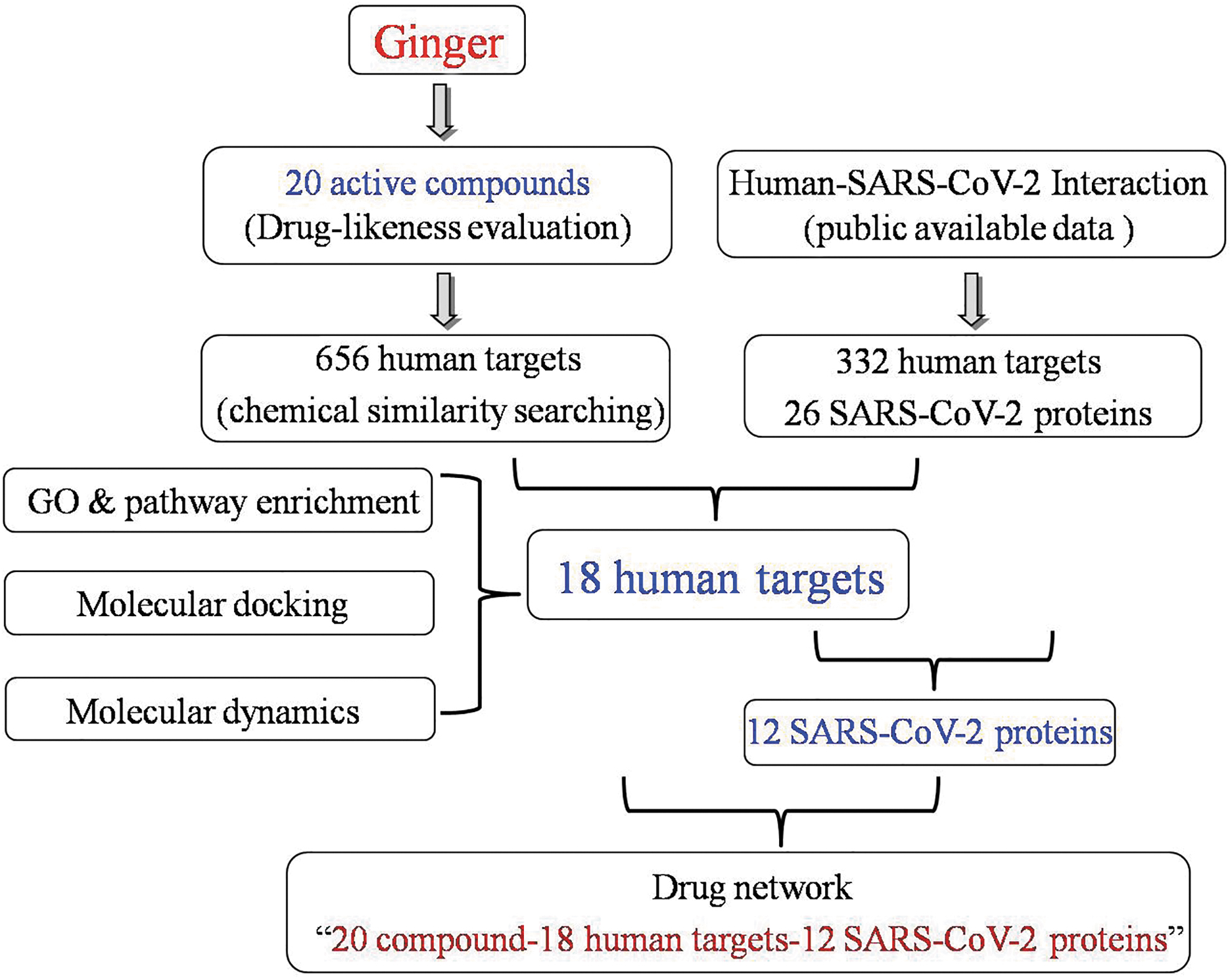
Workflow of the processing.
Materials and Methods
Drug-likeness estimations by the TCMSP server
Drug-likeness (DL) is a qualitative calculation used in the early stages of the drug research process to identify a compound with interesting pharmacokinetics and pharmaceutical attributes and to estimate how “drug-like” it is in comparison with some known or approved drugs.
18,19
The TCMSP server (
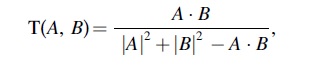
where A is the molecular descriptors of herbal ingredients and B is the average molecular properties of all molecules in the DrugBank database. 19 In this study, compounds with DL ≥0.18 (the average DL of all molecules in the DrugBank database is 0.1819) were chosen as candidate compounds for further research.
For potentially effective compounds, important absorption, distribution, metabolism and excretion properties, such as human oral bioavailability (OB), Caco-2 permeability (Caco-2), blood–brain barrier (BBB), and AlogP (Lipinski's rule of five) were also obtained from the TCMSP database.
Target identification using chemical similarity searching
The targets of many active natural products have not been explored, and this greatly hinders the development of new drugs.
21
SwissTargetPrediction (
Identification of targets of ginger according to the SARS-CoV-2 protein interaction map
Data mapping maps relevant protein targets to diseases, which can be a useful analytics tool for drug target identification. 24,25 SARS-CoV-2-human PPIs were thoroughly researched by Gordon et al. 26 They cloned 26 of the 29 SARS-CoV-2 proteins, transfected them into human HEK-293T/17 cells for more than 38 h, and then performed affinity purification mass spectrometry to identify interactions between SARS-CoV-2 and human proteins. 26 The raw data for the SARS-CoV-2 protein interaction map are freely available and can be downloaded from the PubMed database. 26 For data mapping, the potential targets of ginger that overlap with human proteins that physically interact with SARS-CoV-2 were analyzed using a Venn diagram and further researched.
PPIs, enrichment analysis, and network construction
A better understanding of PPIs can help to elucidate their mechanisms and gain insight into the nature of cellular activities.
27
GeneMANIA (
For meaningful biological functional annotation of these potential targets, GO enrichment and KEGG pathway enrichment were analyzed on the OmicShare platform. Finally, a network comprising ginger compounds, human protein targets, and SARS-CoV-2 proteins was built to offer a simple view of the anti-SARS-CoV-2 mechanisms of ginger.
Molecular docking verification
Knowledge of ligand–protein complexes is essential to grasp the mechanism of action of a drug.
28
AutoDock Vina is widely used to analyze how a small molecule binds to a protein.
28,29
The crystal structures of protein targets were downloaded from the RCSB protein databank (
Molecular dynamics simulation
To assess the stability of the binding between potentially effective compounds of ginger and sigma nonopioid intracellular receptor 1 (SIGMAR1) and to validate the molecular docking results, molecular dynamics (MD) simulations were performed for 150 ns in AMBER software. The process and parameter settings were similar to those performed previously. 30 The root mean square deviation (RMSD), root mean square fluctuation (RMSF), and binding free energies (△G bind) of the complexes were computed for further analysis.
Results
Screening active compounds of ginger
A sum of 265 compounds from ginger were gathered from the TCMSP database. There were 19 active compounds with DL ≥0.18. It should be noted that these included two isomers of hexahydrocurcumin (MOL002468 and MOL006126 in Table 1). 6-Gingerol, with DL = 0.16, is a characteristic chemical constituent of ginger that could not be disregarded. Thus, 20 active compounds of ginger, representing 7.54% of all compounds collected, were selected for further research (Table 1). Furthermore, OB, Caco-2 permeability, BBB, and AlogP properties are listed in Table 1.
Drug-Likeness Values of Candidate Active Compounds of Ginger
BBB, blood–brain barrier; DL, drug-likeness; OB, oral bioavailability.
Target fishing by SwissTargetPrediciton
Potential protein targets of the active compounds of ginger were screened by SwissTargetPrediction. We identified 656 putative targets after deletion of duplicates and manual checking (Fig. 2). There were 332 high-confidence protein interactions after mining the human–SARS-CoV-2 interaction map data downloaded from the PubMed database (Fig. 2). Eighteen targets overlapped for 20 active compounds of ginger for SARS-CoV-2 (Fig. 2 and Table 2).
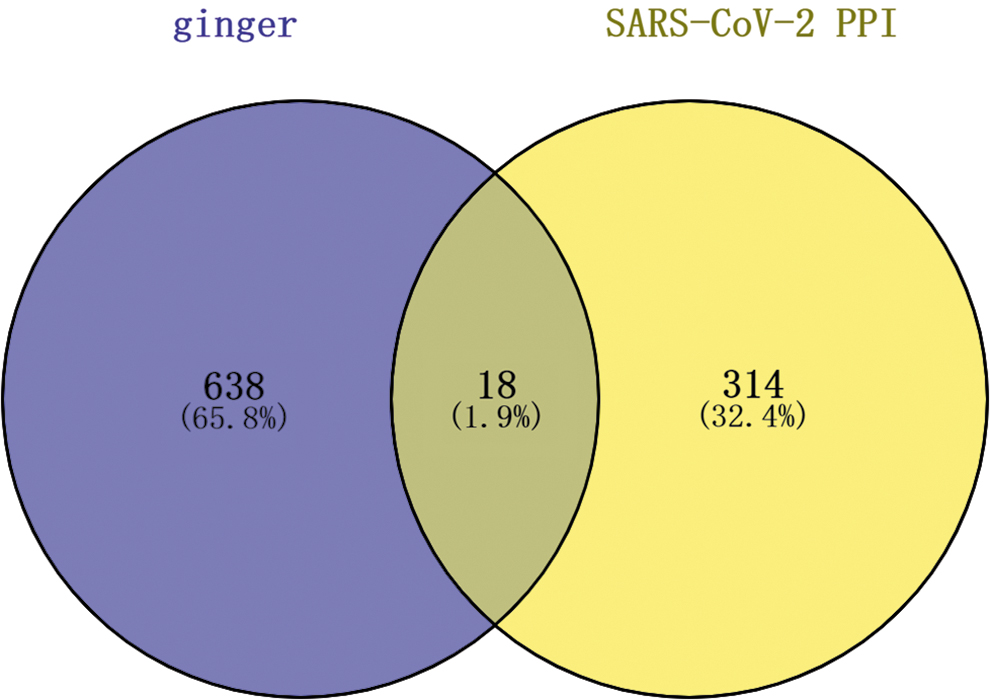
Potential anti-SARS-CoV-2 targets of active compounds of ginger. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Protein Targets of Active Compounds of Ginger That Elicit Anti-Syndrome Coronavirus 2 Effects
We chose two key proteins, SIGMAR1 and histone deacetylase 2 (HDAC2), which are hub proteins and discussed later in the Results section, to validate the SwissTargetPrediction method (Table 2). A search of the DrugBank database revealed that the approved drugs, dextromethorphan and vorinostat, act on SIGMAR1 and HDAC2, respectively. We uploaded the structure files of these two drugs to the SwissTargetPrediction servers independently. SIGMAR1 and HDAC2 were ranked nos. 2 and 3 in the validation results, respectively (Table 3), demonstrating that the method is accurate and reproducible.
Drug and Target Information for Method Validation
GeneMANIA analysis
In terms of the relationships between the 18 targets and their proteins of interest, 77.24% are physical interactions and 19.11% are coexpression (Fig. 3). These accounted for the vast majority of interactions, indicating that these proteins have similar biological functions.
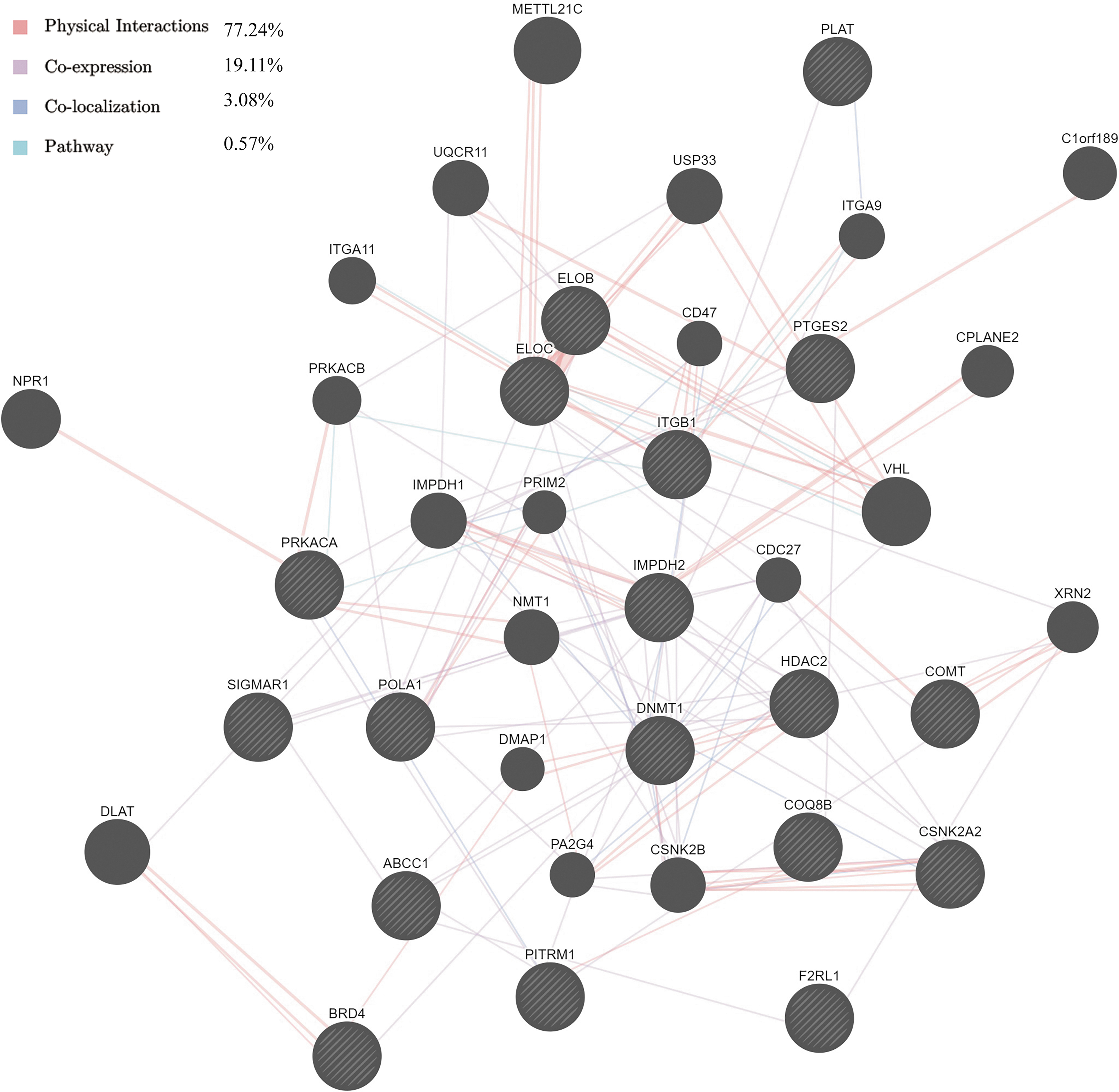
Analysis of the target network by GeneMANIA. Black nodes, protein targets; colored lines, different interactions.
Enrichment analysis and network construction
The top 3 biological processes in GO enrichment analysis were “multi-organism process,” “cellular response to stress,” and “transcription elongation from RNA polymerase II promoter” (Fig. 4a). The 18 proteins may engage in many KEGG pathways. Among these KEGG pathways, the top 3 were “Pathways in cancer” “MicroRNAs in cancer,” and “Longevity regulating pathway-multiple species” (Fig. 4b).
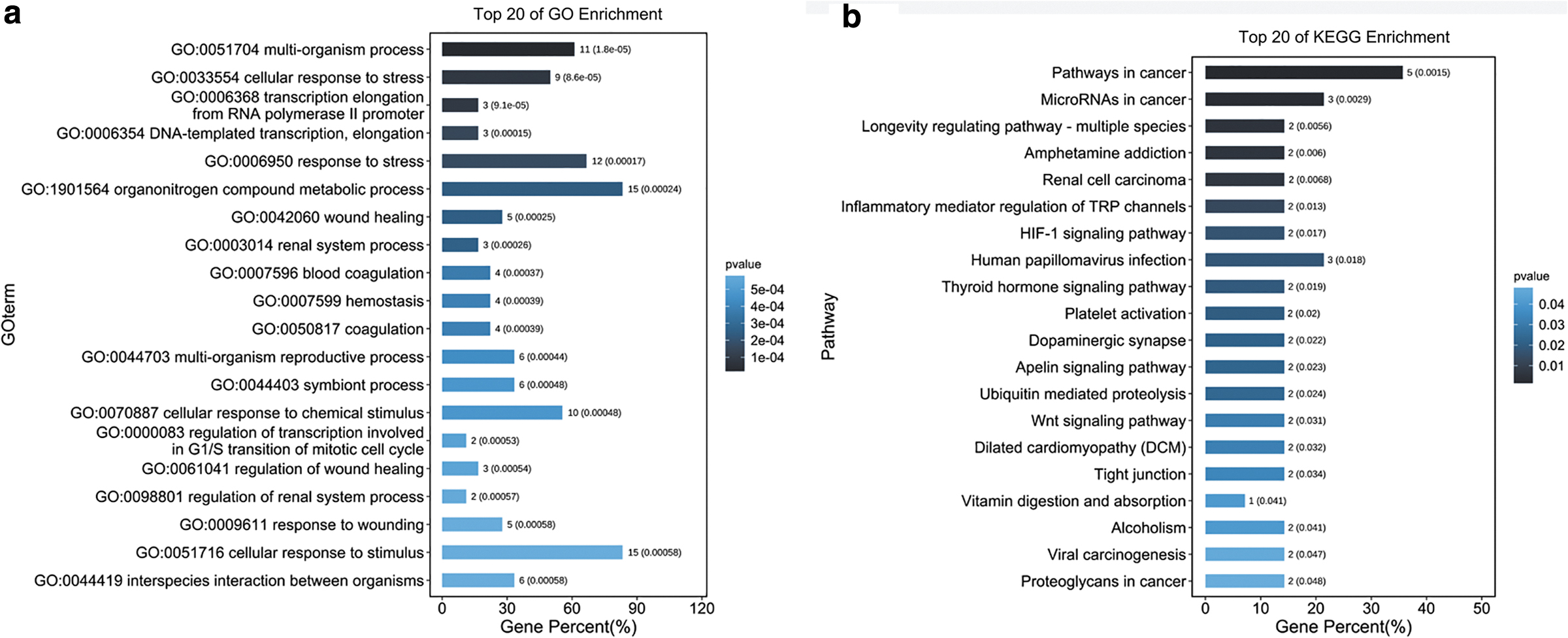
Enrichment analysis of targets.
Of the 332 human targets that interact with SARS-CoV-2 proteins, 62 are targets of 69 drugs, investigational drugs, and preclinical molecules. 26 Accordingly, 13 of the 18 overlapping human targets of ginger may be druggable (Fig. 5). There were 332 high-confidence interactions with 26 SARS-CoV-2 proteins, and we also found that 18 human targets of ginger can act to 12 SARS-CoV-2 proteins (Table 4).
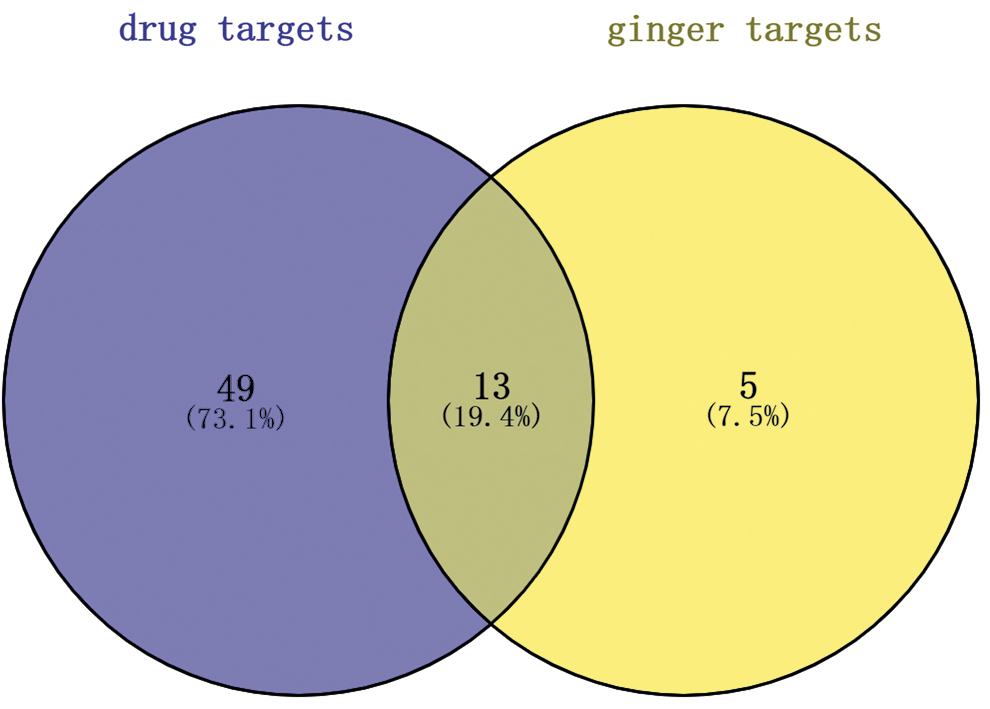
The druggable targets of ginger to elicit anti-SARS-CoV-2 effects.
Syndrome Coronavirus 2 Proteins That Interact with Human Targets of Active Compounds of Ginger
SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Based on these results, we constructed an interaction network comprising 20 ginger compounds, 18 human targets, and 12 SARS-CoV-2 proteins to offer a systemic and simple outline of the mechanism of action for ginger against SARS-CoV-2 (Fig. 6). According to node degree in the network, SIGMAR1 and HDAC2 may be druggable hub targets (Fig. 6).
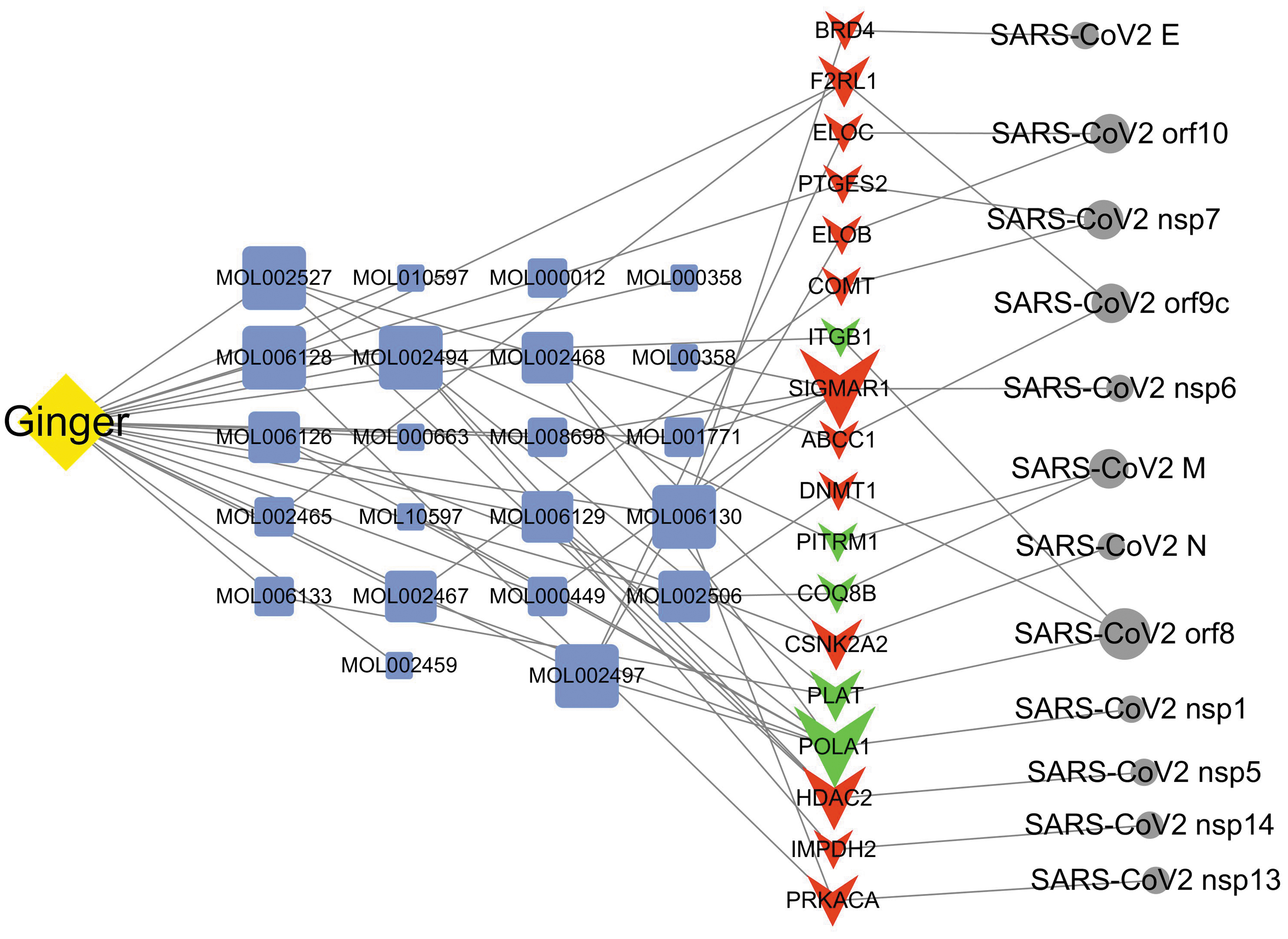
The ginger–compound–target–SARS-CoV-2 protein network. Yellow diamond, ginger; blue box, compounds; green triangles, target proteins; red triangles, druggable target proteins; and gray circles, SARS-CoV-2 proteins. The node size is associated with the node degree.
Molecular docking verification
SIGMAR1 had the top node degree in the interaction network for all 18 targets (Fig. 6), and we therefore chose it (PDB code 5HK1, with the original ligand PD144418) for molecular docking verification. As illustrated in Supplementary Figure S1, the redocking pose of PD144418 fitted as well as the conformation in cocrystal structure, with a RMSD of only 0.57 Å for nonhydrogen atoms. This indicated that the docking procedure and parameters were suitable for further study.
Among the five active compounds of ginger that can interact with SIGMAR1, the docking energy of the lowest state of the 8-gingerdione–SIGMAR1 complex was −8.0 kcal/mol, while the docking energy of the original ligand-SIGMAR1 was −10.1 kcal/mol (Table 5).
Binding Energy of Active Compounds of Ginger to Sigma Nonopioid Intracellular Receptor 1
The 2D interactions of the five active compounds of ginger that can interact with SIGMAR1 are shown in Figure 7. The 8-Gingerdione can form hydrogen bonds with residue Tyr103 of SIGMAR1 (Fig. 7a). Dihydrocapsaicin can form hydrogen bonds with residues Tyr103 and Glu172 of SIGMAR1 (Fig. 7b). Therefore, the docking energy of both compounds may be similar to that of the original ligand PD144418. Stigmasterol and clionasterol can form hydrogen bonds with residue Ser117 of SIGMAR1 (Fig. 7c, d). Beta-sitosterol can form an unfavorable bump with Tyr120 of SIGMAR1, and therefore the docking energy is relatively high (Fig. 7e and Table 5). Figure 7f shows the interaction of the original ligand PD144418 with SIGMAR1. Other interaction information, such as van der Waals bonds, is provided in Figure 7. Based on these results, 8-gingerdione and dihydrocapsaicin may preferentially interact with SIGMAR1 (Table 5 and Fig. 7).
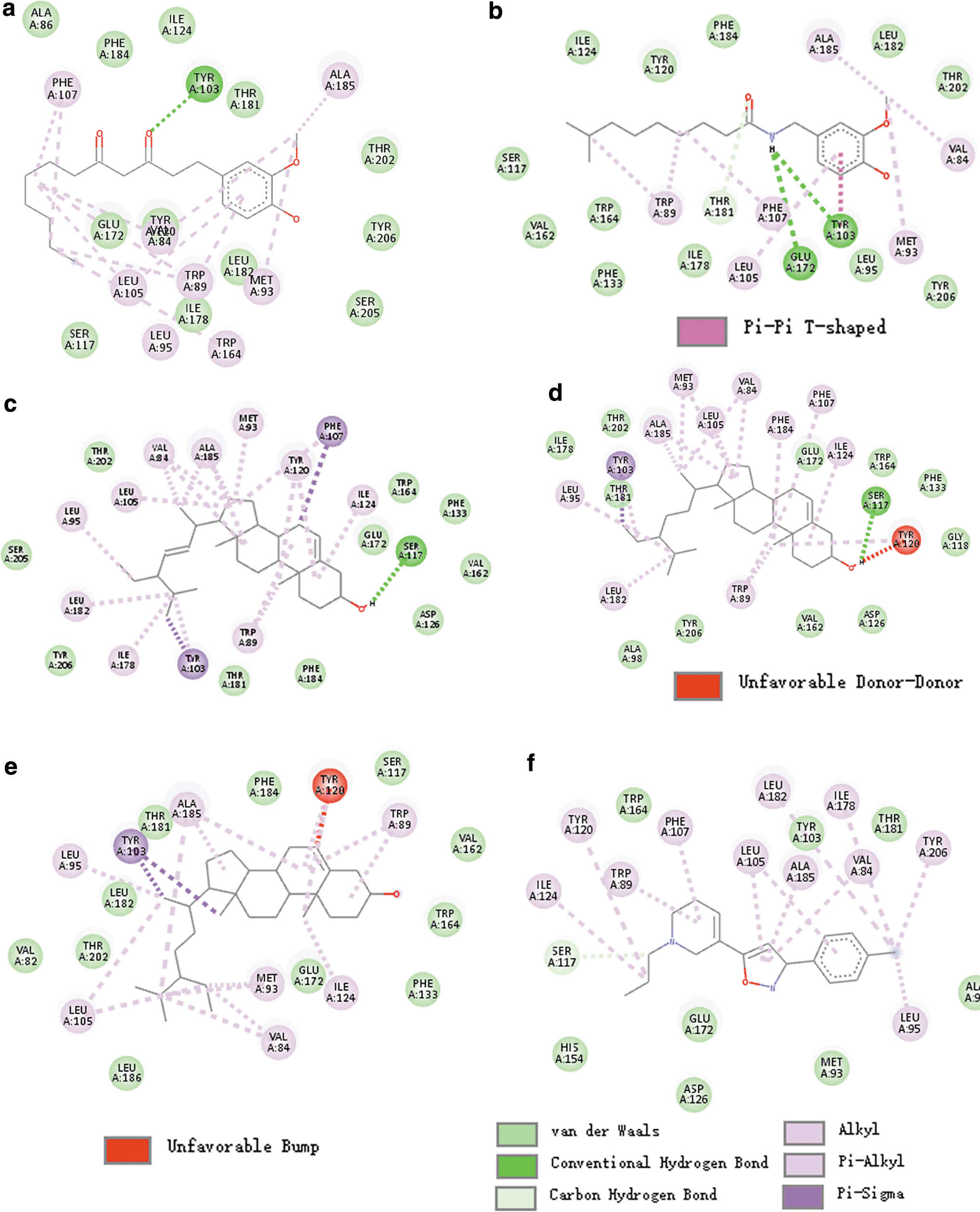
Molecular interactions between SIGMAR1 and active compounds of ginger.
MD stimulation
Generally, the RMSD did not fluctuate significantly during the simulation period (Fig. 8a, b). The RMSD values of the PD144418–SIGMAR1 complex ranged from 1.555 to 4.369 Å (Fig. 8a, b). For all five compounds, the conformation of MOL006130 and MOL008698 were closer to the conformation of PD144418 (Fig. 8a). The residues with higher RMSF values may be more flexible. As shown in Figure 8c and d, the active site residues, such as Glu172, Met93, Leu95, Leu105, Phe107, Ile124, Trp164, and Glu172, had small RMSF values (less than 1.0 Å). This indicated that the binding pocket in the active site of SIGMAR1 was stable.

The predicted binding free energies were less than zero, suggesting that the binding interactions were spontaneous (Table 6). The predicted binding free energy of the PD144418–SIGMAR1 complex was −211.61 kcal/mol, whereas the binding energies of MOL00613 and MOL008698 to SIGMAR1 were −210.58 and −198.72 kcal/mol, respectively, indicating that they had better binding affinities (Table 6). Furthermore, nonpolar solvation, van der Waals, and electrostatic interactions also contributed favorably to ligand binding (Table 6). Overall, the two compounds appeared to bind tightly to SIGMAR1.
Binding Free Energy of Complexes Along with Individual Energy Component Contributions (kcal/mol)
Contribution to the free energy of binding from the van der Waals energy. 30
Contribution to the free energy of binding from the electrostatic energy. 30
Contribution to the free energy of binding from the polar salvation energies. 30
Contribution to the free energy of binding from the nonpolar solvation energies. 30
Free energy of binding.
Discussion
Numerous studies have revealed that ginger has antiviral effects. Fresh ginger elicits inhibitory effects on human respiratory syncytial virus in human respiratory tract cell lines. 31 An aqueous extract of ginger exhibits antiviral activity against Avian influenza virus H9N2 in chick embryos 32 and Chikungunya virus in Vero cells. 33 Gingerenone-A, an important compound present in ginger, inhibits the replication of influenza A (H5N1) virus in 293T cells and experimental mice. 34 Furthermore, ginger silver nanoparticles have anti-SARS-CoV-2 activity with an IC50 value of 0.034 μg/mL in Vero cells. 35 Therefore, ginger can potentially inhibit SARS-CoV-2, and its pharmacological mechanisms are worthy of systematic exploration.
The interactions of 332 human proteins with SARS-CoV-2 can improve the understanding of virus infection and morbidity. 26 The active compounds of ginger may act on 18 of these 332 human proteins to elicit anti-SARS-CoV-2 effects (Fig. 2 and Table 2). SIGMAR1 and HDAC2 may be hub proteins in the pharmacological network (Fig. 6).
SIGMAR1 is a widely expressed signaling chaperone protein and plays miscellaneous roles in cellular survival. 36 It interacts with nonstructural protein (NSP) 6 and open reading frame (ORF) 9c of SARS-CoV-2, functions in lipid remodeling and the stress response of the endoplasmic reticulum, and affects viral replication. 26 A clinical trial showed that fluvoxamine, which has a high affinity for SIGMAR1, can prevent clinical deterioration of adult outpatients with symptomatic COVID-19. 37 Five active compounds of ginger (8-gingerdione, dihydrocapsaicin, stigmasterol, clionasterol, and beta-sitosterol) may interact with SIGMAR1, indicating that ginger may suppress replication of SARS-CoV-2 through SIGMAR1 (Fig. 6 and Table 5).
HDAC2 is an important part of the host innate antiviral reaction to influenza A virus. 38 The SARS-CoV-2 protein NSP5 interacts with HDAC2, inhibits the transport of HDAC2 into the nucleus, and potentially affects the host immune response. 26 Ginger downregulates HDAC2 and HDAC3 expression in Caco-2 cells. 39 The 6-Shogal, one of the most bioactive compounds of ginger, inhibits HDACs and induces heat shock protein 70 expression in primary rat astrocytes to suppress lipopolysaccharide-induced inflammation. 40 Therefore, ginger may elicit anti-SARS-CoV-2 effects through HDAC2. Hence, ginger exerts inhibitory effects on SARS-CoV-2 through multiple target proteins (Table 2).
Our results showed that ginger affects 12 of 26 SARS-CoV-2 proteins (Fig. 5 and Table 4). This finding is supported by many in silico studies. For example, ginger is a candidate herb for the treatment and prevention of COVID-19 based on a report showing that its compounds bind to the SARS-CoV-2 3CL protease (Mpro) and papain-like protease using the TCMD 2009 database. 41 Gingerol can bind to NSP15 protein. 42 The 6-Gingerol also has a high binding affinity for spike protein, with binding energy of −4.177 kcal/mol. 43 Gingerenone-A may dock well with five SARS-CoV-2 proteins (Mpro, ADP ribose phosphatase, NSP14, NSP16, and PLpro). 35 The 8-Gingerol and 10-gingerol present in ginger have better GlideScores values (−5.88 and −5.72 kcal/mol, respectively) for the SARS-CoV-2 main protease than hydroxychloroquine (−5.47 kcal/mol). 44 Gma-miR-166m, which is abundant in ginger, can target ORF8, while gma-mir-4995 of ginger can target spike protein. 45 Six compounds (10-paradol, 8-paradol, scopoletin, 10-shogaol, 8-gingerol, and 10-gingerol) are powerful inhibitors of angiotensin-converting enzyme 2, which is a key protein for SARS-CoV-2 entry. 43
Pharmacological experiments have been conducted to demonstrate the anti-SARS-CoV-2 effect of ginger. Ginger exosome-like nanoparticle miRNA (aly-miR396a-5p) inhibits mouse lung inflammation induced by NSP12 protein and inhibits the cytopathic effect in Vero E6 cells by suppressing the expressions of viral S and NSP12 proteins. 46 Thus, ginger compounds may directly affect SARS-CoV-2 virus proteins.
Molecular docking verification may offer information on the binding affinity and molecular structure recognition for a compound–protein complex. 28 Five ginger compounds can interact with SIGMAR1. The docking energy of 8-gingerdione and dihydrocapsaicin is lower and similar to that of the original ligand PD144418, while that of beta-sitosterol is much higher (Table 5). This was confirmed using MD stimulation (Table 6). Residue Tyr103 of SIGMAR1 can form a hydrogen bond with the ligand and is responsible for a large proportion of the binding affinity. 47 Both 8-gingerdione and dihydrocapsaicin can form a hydrogen bond with Tyr103 (Fig. 7a, b), consistent with the docking energy results (Table 5). Thus, both may preferentially interact with SIGMAR1.
Meanwhile, beta-sitosterol may form an unfavorable bump with Tyr120, indicating that the docking energy is much higher (Table 5 and Fig. 7e). The molecular docking and MD energy results (Tables 5 and 6) are consistent with the interaction information (Fig. 7). Thus, both 8-gingerdione and dihydrocapsaicin should be further investigated as promising compounds of ginger to target SARS-CoV-2.
In conclusions, based upon a SARS-CoV-2-human PPI map, we used network pharmacology tools to reveal the potential anti-COVID-19 targets for ginger. We found that the antiviral activity of ginger may involve 20 active compounds, 18 human targets, and 12 SARS-CoV-2 proteins. These constitute an interaction network in which SIGMAR1 and HDAC1 may be hub proteins. In addition, molecular docking showed that 8-gingerdione and dihydrocapsaicin may preferentially interact with SIGMAR1. Therefore, ginger targets multiple human and SARS-CoV-2 proteins to exert pharmacological effects on COVID-19. Nevertheless, more experiments are needed to further confirm these findings.
Data Availability Statement
The raw data are openly available from the authors.
Footnotes
Authors' Contributions
L.H.Z. and S.J.C. designed the study; S.J.C. performed and analyzed the data; S.J.C. wrote the article; and L.H.Z. revised the article. All authors have approved the final version.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The study was funded by the Ningbo Natural Science Foundation (2023J308).
Supplementary Material
Supplementary Figure S1