
Abstract
This study investigates the protective effects of CGK012 [(7S)-(+)-cyclopentyl carbamic acid 8,8-dimethyl-2-oxo-6,7-dihydro-2H,8H-pyrano[3,2-g]chromen-7-yl-ester], a small-molecule inhibitor targeting the Wnt/β-catenin signaling pathway, against inflammatory responses elicited by lipopolysaccharide (LPS). The study evaluated the influence of CGK012 on heme oxygenase (HO)-1, cyclooxygenase (COX)-2, and inducible nitric oxide synthase (iNOS) expressions in LPS-stimulated human endothelial cells. It examined its effects on iNOS, tumor necrosis factor (TNF)-α, and interleukin (IL)-1β in LPS-challenged mice. CGK012 treatment resulted in increased HO-1 production, inhibited nuclear factor-kappa B activation, and decreased the levels of COX-2/PGE2 and iNOS/NO. Additionally, CGK012 reduced signal transducer and activator of transcription-1 phosphorylation and facilitated Nrf2 nuclear translocation and binding to antioxidant response elements, culminating in reduced IL-1β production in LPS-exposed human umbilical vein endothelial cells. Notably, the inhibitory effect of CGK012 on iNOS/NO was reversed upon HO-1 knockdown via RNA interference. In vivo, CGK012 markedly attenuated iNOS expression in lung tissue and decreased TNF-α levels in bronchoalveolar lavage fluid. These findings underscore the anti-inflammatory potential of CGK012, suggesting its therapeutic promise for conditions characterized by pathological inflammation.
INTRODUCTION
Heme oxygenase-1 (HO-1) is a critical enzyme for mitigating damage associated with inflammation and reactive oxygen species accumulation, particularly in severe pathological conditions, including acute autoimmune reactions, pulmonary diseases, and malignancies. 1,2 HO-1 exerts anti-inflammatory effects by inhibiting the production of key proinflammatory cytokines and mediators, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6. 2 Prior studies have underscored the protective effects of HO-1 in animal models, specifically demonstrating its capacity to shield mice from acute septic inflammation and other inflammation-associated vascular disorders. 2,3 The Keap1-Nrf2-antioxidant response element (ARE) pathway plays an indispensable role in the cellular response to oxidative stress. It modulates inflammatory processes by regulating detoxifying and antioxidant genes essential for degrading carcinogens and other toxins. 4 Nrf2 is integral to cytoprotective signaling, which establishes the Nrf2-ARE pathway as a pivotal target for intervention in inflammation-related pathologies. 4,5 Additionally, the signal transducer and activator of transcription (STAT)-1 and nuclear factor (NF)-κB pathways are crucial to inflammatory signaling, 6,7 with STAT1 and NF-κB activation driving the production of proinflammatory mediators that attract immune cells to inflamed tissue. 6,7 Dysregulation of these pathways can result in chronic inflammation and tissue damage, thereby underscoring the therapeutic potential of targeting STAT-1 and NF-κB in inflammatory diseases.
Lung inflammation presents a significant clinical challenge, often manifesting as proliferative edema, hypoxia, and the formation of neutrophil extracellular traps within pulmonary tissue. 8 Lipopolysaccharide (LPS) is a primary contributor to respiratory pathologies. It promotes the release of proinflammatory mediators through the activation of transcriptional regulators. This mechanism sustains inflammatory responses, which are implicated in conditions such as vascular dysfunction, asthma, chronic obstructive pulmonary disease, and cystic fibrosis. 8
Furthermore, activation of the Wnt/β-catenin pathway has been correlated with increased expression and release of HMGB1 in intestinal tumorigenesis. 9 A novel pyranocoumarin compound, CGK012 [(7S)-(+)-cyclopentyl carbamic acid 8,8-dimethyl-2-oxo-6,7-dihydro-2H,8H-pyrano[3,2-g]chromen-7-yl-ester], has demonstrated efficacy in attenuating Wnt/β-catenin signaling by inhibiting β-catenin transcription and activation in the presence of Wnt3a-CM. 10 This attenuation has been associated with decreased proliferation in multiple myeloma cells by reducing intracellular β-catenin levels. 10 CGK012 induces β-catenin phosphorylation at Ser33/Ser37/Thr41 through a glycogen synthase kinase-3β (GSK-3β)-independent pathway, promoting proteasomal degradation and thus lowering intracellular β-catenin levels. Additionally, CGK012 consistently downregulates β-catenin and suppresses the expression of downstream targets, including cyclin D1, c-myc, and axin-2, in RPMI-8226 multiple myeloma cells. 10 The present study aims to evaluate the effects of CGK012 on HO-1 and inflammatory mediators, including TNF-α, IL-1β, and nitric oxide (NO), in human umbilical vein endothelial cells (HUVECs) in vitro, as well as on the histological architecture of LPS-challenged mice in vivo. The overarching objective is to elucidate the potential role of CGK012 in modulating HO-1 signaling pathways and suppressing inflammatory cytokine expression, thereby assessing its viability as a therapeutic agent in inflammatory diseases.
MATERIALS AND METHODS
Cell culture and materials
HUVECs (C2517AT25; Cambrex BioScience, Charles City, IA, USA) were maintained according to a previously established protocol. 11 The relevance of using HUVECs lies in their critical role in endothelial dysfunction, a key factor in systemic inflammation and lung injury. As such, they provide an appropriate model for investigating inflammation pathways. 12,13 HUVECs enable a mechanistic exploration of key signaling pathways, including NF-κB, STAT1, and Nrf2, which are pivotal in regulating inflammation and oxidative stress during lung injury. 14 Furthermore, HUVECs, being of human origin, offer enhanced translational value for human inflammatory conditions and are widely recognized as a standardized model for studying endothelial function and inflammatory responses. 12,13 Dexamethasone (Dex, positive control), LPS (from Escherichia coli), penicillin G, streptomycin, and dimethyl sulfoxide (DMSO) were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Human HO-1 short interfering RNA (siRNA) (sc-35554) and control siRNA (sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). HUVECs from passages 3–5 were seeded at a density of 1 × 105 cells per 35 mm dish and starved overnight prior to enzyme-linked immunosorbent assay (ELISA) analysis. For the experimental treatments, some cells were exposed to LPS (1 μg/mL for 6 h) followed by CGK012 treatment for an additional 6 h, while others received CGK012 treatment for 6 h without prior LPS exposure to assess HO-1 expression levels.
Method for the synthesis of (7S)-(+)-cyclopentyl carbamic acid 8,8-dimethyl-2-oxo-6,7-dihydro-2H,8H-pyrano[3,2-g]chromen-7-yl-ester (CGK012)
The synthetic procedure for CGK012 is depicted in Scheme 1. To a solution of (+)-decursinol (1 g, 4.06 mmol, 1 eq) in anhydrous toluene (300 mL), cyclopentyl isocyanate (0.67 g, 6.09 mmol, 1.5 eq), triethylamine (0.74 g, 7.31 mmol, 1.8 eq), and 4-dimethylaminopyridine (0.29 g, 2.44 mmol, 0.6 eq) were added sequentially. The reaction mixture was then stirred at 110°C for 24 h. After confirming the completion of the reaction by TLC, the mixture was cooled to room temperature, filtered through celite, and the filtrate was washed with toluene. The combined organic layers were then subjected to three successive washes with 300 mL of 3N HCl aqueous solution. The acid-treated organic layers were further washed with 300 mL of a saturated K2CO3 aqueous solution followed by 300 mL of water. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The resulting solid was subjected to crystallization using n-pentane, followed by filtration to afford CGK012 as an ivory-white solid with a yield of 98.4%. The compound’s characteristics were as follows: mp 162 °C; [α]
Synthesis of CGK012.
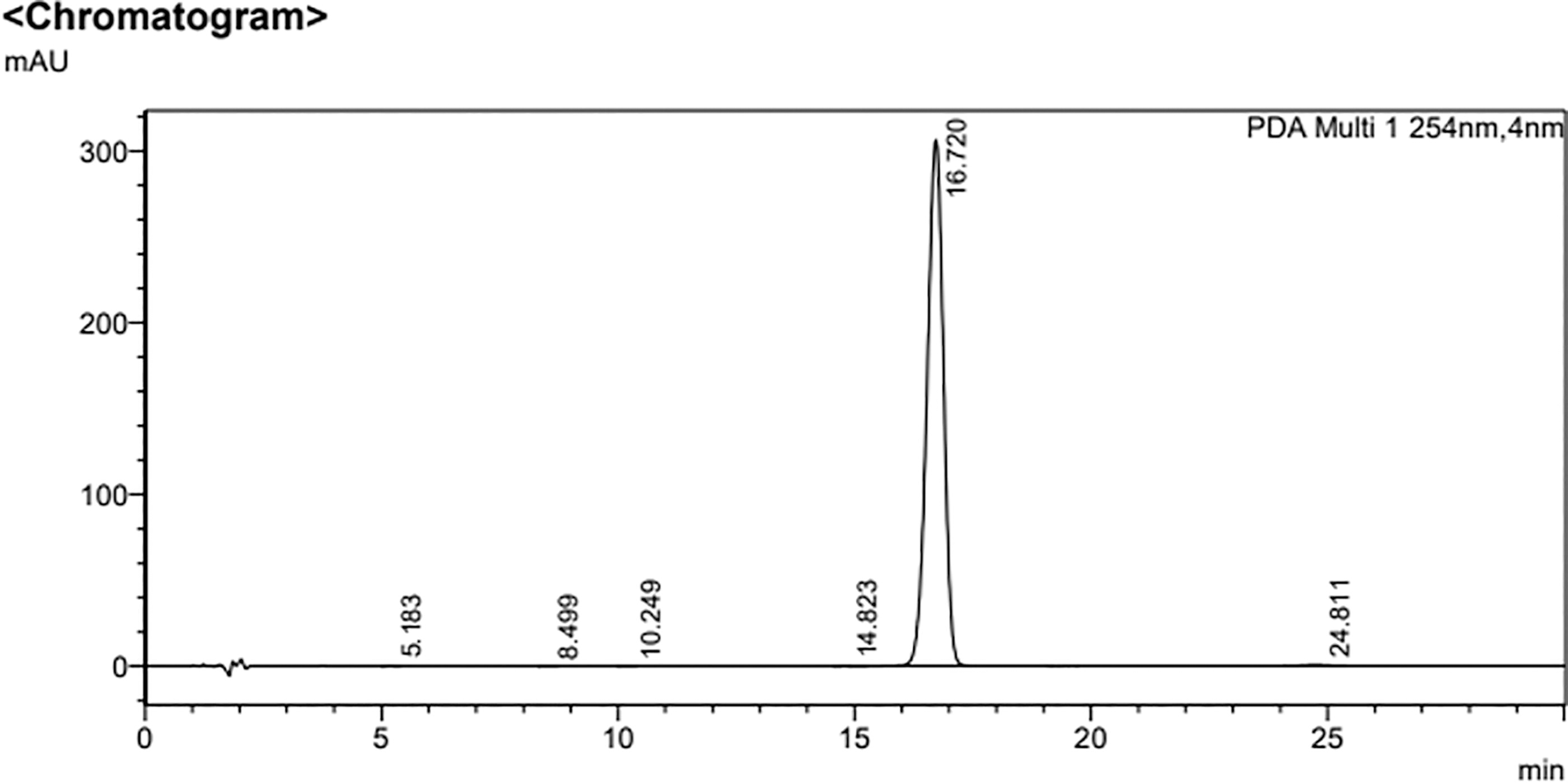
HPLC chromatogram of CGK012. The purity of CGK012 was determined as 99.7% by analytical HPLC on a shim-pack GIS-ODS 5 μM C18 column (150 × 4.6 mm). The mobile phase conditions were set from an isocratic solvent system; A [0.1% phosphoric acid (v/v) in water] and B (acetonitrile). The detailed isocratic elution was as follows: 0–30 min, 45% B. The flow rate of the mobile phase was 1.0 mL/min. The wavelength was set at 254 nm. The column temperature was 40°C. The injection volume was 20 µL.
Mouse model of LPS-induced lung injury
Male C57BL/6 mice (6–7 weeks old, mean weight 27 g) were obtained from Orient Bio Inc. (Seongnam, Republic of Korea). Prior to initiating experimental procedures, the mice were acclimatized for 12 days to ensure adaptation to laboratory conditions, in accordance with established protocols. 15,16 LPS (15 mg/kg) dissolved in 0.2% DMSO as a vehicle was administered intraperitoneally using a 28-gauge needle. Six hours post-LPS administration, CGK012 was injected intravenously at doses ranging from 0.06 to 0.53 mg/kg (n = 5/group). All experimental procedures were conducted with approval from the Animal Care Committee at Kyungpook National University (IRB No. KNU 2022-112). Bronchoalveolar lavage fluid (BALF) was collected following intratracheal administration of phosphate-buffered saline (PBS) using moderate suction, and the samples were centrifuged at 2000g. for 10 min at 4°C. The supernatant was stored at −80°C for subsequent analyses.
ELISA
The effect of CGK012 on HUVECs was evaluated by stimulating the cells with LPS (1 μg/mL) for 6 h, followed by treatment with CGK012 for an additional 6 h. Additionally, separate cell groups were treated with CGK012 without prior LPS stimulation to assess HO-1 concentrations. The phosphorylation levels of STAT1 were determined using ELISA kits (ab126455, Abcam, Cambridge, MA, USA). Prostaglandin E2 (PGE2), HO-1, IL-1β, TNF-α, and inducible nitric oxide synthase (iNOS) levels were quantified in supernatants collected after centrifugation of the cell cultures by using ELISA kits from R&D Systems.
Cell viability assay
Cell viability was assessed using the MTT assay, as previously described. 17,18 HUVECs were seeded at a density of 5 × 103 cells per well in 96-well plates and treated with CGK012 for 48 h. Following treatment, the cells were washed and incubated with MTT solution (1 mg/mL, 100 μL/well) for an additional 4 h. The resulting formazan crystals were solubilized by adding 150 μL of DMSO, and absorbance was measured at 540 nm using a spectrophotometer (Tecan, Austria GmbH). Cell viability was expressed as a percentage relative to untreated control cells (100%).
Nitrite levels
Nitric oxide production was evaluated by measuring nitrite (NO2 –) concentrations in the cell culture medium. Equal volumes of Griess reagent (ab234044, Abcam) were added to the supernatant, followed by a 15-min incubation at room temperature. The absorbance of the resulting solution was measured at 540 nm using a spectrometer. The assay was repeated in triplicate to ensure reproducibility and reliability of the results.
Intracellular fractionation, coimmunoprecipitation, and immunoblotting
Following cell harvesting, the supernatants were separated by centrifugation. Cytosolic and nuclear extracts were subsequently isolated using a protocol previously established in the literature. 19 Immunoblotting was performed using antibodies against iNOS, cyclooxygenase (COX)-2, lamin B, Keap1, Nrf2, and β-actin, all obtained from Santa Cruz Biotechnology (CA, USA). Lamin B and β-actin were used as loading controls for the nuclear and cytosolic extracts, respectively. The Keap1-Nrf2 interaction in HUVECs was analyzed by coimmunoprecipitation using protein A-agarose. Briefly, treated HUVECs were lysed and centrifuged at 4°C to obtain the supernatant, which was incubated with suspended protein A-agarose at 4°C to reduce non-specific binding. After centrifugation for 5 min, the supernatant was incubated with either anti-Nrf2 or control IgG for 1 h at 4°C, followed by the addition of protein A-agarose. The mixture was subjected to overnight shaking, after which the immunoprecipitates were collected by centrifugation. The supernatant was discarded, and the pellet was washed three times with lysis buffer. The pellets were then resuspended in SDS loading buffer and heated at boiling temperature for 10 min. The precipitated proteins were subsequently analyzed by Western blot using the indicated antibodies.
Quantitative real-time polymerase chain reaction
Total RNA was extracted using TRI Reagent (Invitrogen), followed by reverse transcription using a 20 µL reaction mixture containing 0.5 mg/µL of the oligo (dT)-adapter primer (Invitrogen) and M-MLV reverse transcriptase (Invitrogen), performed in a PX2 Thermal Cycler (Thermo Scientific). The mRNA expression levels of iNOS, COX-2, Nrf2, and HO-1 were normalized to β-actin as the internal control. The primer sequences used for Real-Time Quantitative Reverse Transcription PCR (qRT-PCR) were as follows: COX-2 forward: 5′-CCC CAT TAG CAG CCA GTT-3′, COX-2 reverse: 5′-CAT TCC CCA CGG TTT TGA-3′; iNOS forward: 5′-GTT CTC AGC CCA ACA ATA CAA GA-3′, iNOS reverse: 5′-GTG GAC GGG TCG ATG TCA C-3′; Nrf2 forward: 5′-TCC TAT GCG TGA ATC CCA AT-3′, Nrf2 reverse: 5′-GCG GCT TGA ATG TTT GTC TT-3′; HO-1 forward: 5′-GGG CTG TGA ACT CTG TCC AAT-3′, HO-1 reverse: 5′-GGT GAG GGA ACT GTG TCA GG-3′; and β-actin forward: 5′-TCG TGC GTG ACA TCA AAG A-3′, β-actin reverse: 5′-CAT ACC CAA GAA GGA AGG CT-3′.
Transfection
Plasmids encoding the NF-κB-luciferase reporter vector, HO-1 siRNA, ARE luciferase reporter vector, and non-targeting control siRNA were transfected into cells using SuperFect transfection reagent (Qiagen, CA, USA). After a 4-h incubation period with the plasmid constructs, the culture medium was replaced with a fresh medium.
ARE luciferase reporter assay
Cells were washed with PBS at room temperature and lysed using the lysis buffer from the dual luciferase assay kit (Promega, WI, USA). Luciferase activity was subsequently quantified with a TD-20/20 luminometer (Turner Designs, CA, USA). Each transfection experiment was performed separately in triplicate. The data were expressed as the ratio of firefly luciferase activity to Renilla luciferase activity.
Histopathological analysis
Five mice received intraperitoneal injections of LPS, followed by intravenous administration of CGK012 (0.53 mg/kg) after a 6-h interval. The mice were then euthanized, and lung tissue samples were collected for histological examination. Tissue sections were stained with hematoxylin and eosin stain (H&E), and histopathological changes were assessed based on previously established criteria. 20 Pulmonary structural scores were assigned according to a grading system ranging from 1 to 4. 19
Evaluation of oxidative stress markers
Superoxide dismutase (SOD) activity was determined using a SOD assay kit (Fluka, Japan) and expressed as U/mg protein. Catalase (CAT) activity was measured with a CAT assay kit (Sigma Aldrich) by monitoring the decomposition rate of H2O2 at 240 nm. Total CAT levels were reported as U/mg protein.
Statistical analysis
Results are presented as means ± standard deviations (SDs) from three independent experiments. Group comparisons were conducted using one-way analysis of variance, followed by Tukey’s post hoc test to identify significant differences. Statistical significance was defined as P < .05.
RESULTS
Effect of CGK012 on iNOS and COX-2 levels in LPS-activated HUVECs
To investigate the influence of CGK012 on the expressions of inflammation-related genes, we analyzed the levels of iNOS and COX-2, both recognized as key proinflammatory markers. HUVECs were stimulated with LPS for 6 h before being treated with various concentrations of CGK012 or 20 μM of Dex for an additional 6 h. Results from qPCR, ELISA, and immunoblotting demonstrated dose-dependent reductions in LPS-induced iNOS and COX-2 expression following treatment with either CGK012 or 20 μM Dex (Fig. 2A–D). Additionally, treatment with CGK012 or Dex decreased the concentrations of associated molecules, PGE2 and NO (Fig. 2E, F). Furthermore, an MTT assay confirmed that CGK012 did not affect cell viability up to concentrations of 100 μM, indicating no cytotoxicity in HUVECs (Fig. 2G). These findings collectively suggest that CGK012 effectively suppresses iNOS production and inhibits LPS-induced NO synthesis.
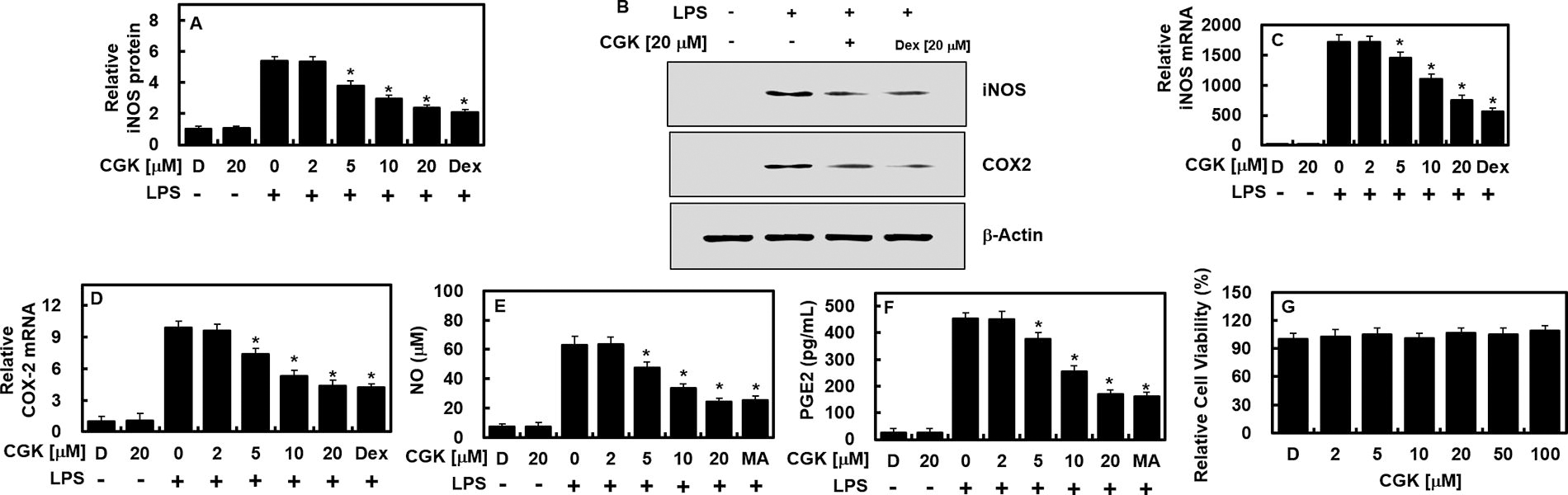
In LPS-stimulated HUVECs, CGK012 administration resulted in decreased levels of both COX-2 and iNOS. Initially, HUVECs were exposed to LPS (1 μg/mL) for 6 h, followed by treatment with varying concentrations of CGK012 or Dex (20 μM) for an additional 6 h. The levels of iNOS protein
Effect of CGK012 on NF-κB activity, STAT-1 phosphorylation, and HO-1 protein levels in LPS-activated HUVECs
We further explored the modulatory effect of CGK012 on NF-κB, a pivotal mediator of inflammation-related gene expression. CGK012 exhibited a dose-dependent inhibition of NF-κB luciferase reporter activity (Fig. 3A). Given the documented role of the JAK/STAT signaling pathway in LPS-induced iNOS and COX-2 production. 21 we assessed CGK012’s effect on STAT-1 phosphorylation and its downstream targets. The results showed that CGK012 suppressed STAT-1 phosphorylation and downregulated its associated targets (Fig. 3B) while markedly increasing HO-1 expression (Fig. 3C).
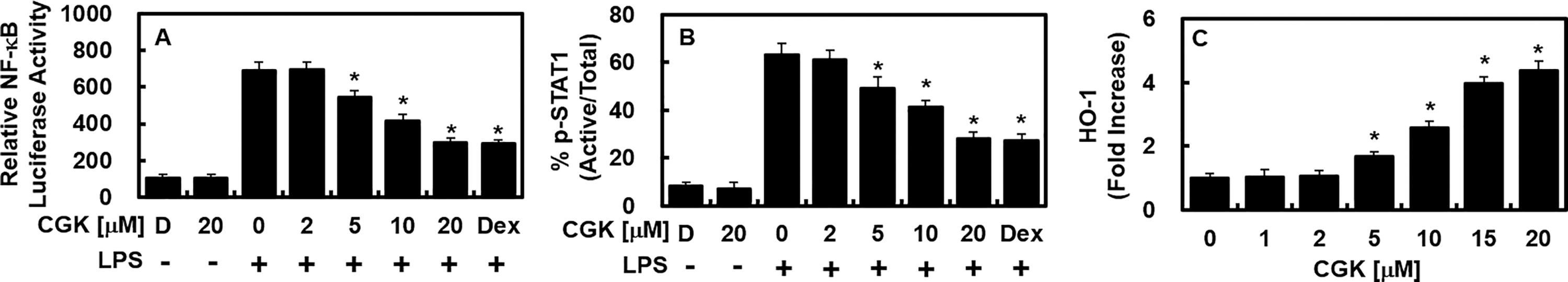
CGK012 suppressed NF-κB and STAT-1 functions while increasing the protein level of HO-1. Following LPS stimulation of HUVECs, treatment with CGK012 or Dex (20 μM) for 6 h at specified concentrations resulted in the suppression of NF-κB and STAT-1 functions, along with an increase in HO-1 protein levels. CGK012 induced elevated levels of HO-1 protein while concurrently inhibiting NF-κB activity and STAT-1 phosphorylation. NF-κB activity was assessed in cells transfected with the NF-κB luciferase reporter vector, whereas STAT-1 phosphorylation was determined using ELISA. Additionally, levels of HO-1 expression from extracted proteins were analyzed via ELISA. The results, representing the means ± SDs from three independent experiments performed in triplicate on three different days, indicate that DMSO treatment (0.2%) served as the vehicle control, labeled as “D.” * P < .05 compared to LPS. ELISA, enzyme-linked immunosorbent assay; HO-1, heme oxygenase-1; NF-κB, nuclear factor-kappa B; STAT, signal transducer and activator of transcription.
Effects of CGK012 on nuclear translocation of Nrf2, ARE reporter activity, and anti-inflammatory responses
To elucidate the role of CGK012 in Nrf2 nuclear translocation and ARE activation, we examined its effects on these processes, given that HO-1 and other antioxidative proteins are Nrf2-dependent. Our findings indicated that CGK012 significantly promoted Nrf2 nuclear translocation and enhanced ARE luciferase reporter activity (Fig. 4A, B). Additionally, to determine whether CGK012’s suppression of iNOS expression was mediated by HO-1 production, HO-1 was silenced using siRNA. This silencing led to restored iNOS and NO levels, mirroring those of untreated cells, thus suggesting that CGK012 augments HO-1 expression, which subsequently reduces iNOS levels (Fig. 4C, D). Finally, CGK012 further demonstrated anti-inflammatory effects by reducing IL-1β expression in LPS-stimulated HUVECs (Fig. 4E).
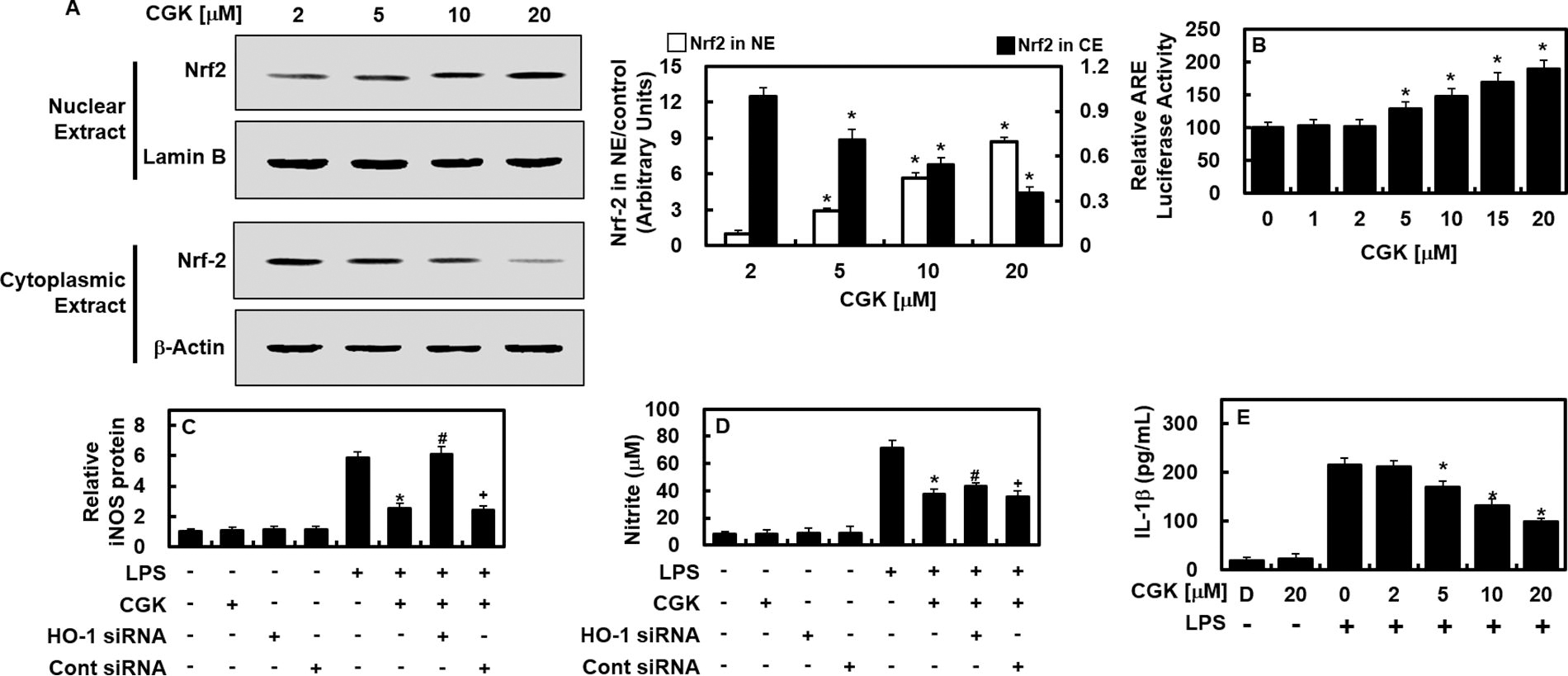
In HUVECs, CGK012 induced the translocation of Nrf2 into the nucleus and demonstrated an anti-inflammatory effect.
Effects of CGK012 on TNF-α and iNOS protein levels in an LPS-mediated lung injury mouse model
The anti-inflammatory effects of CGK012 were evaluated using an LPS-mediated lung injury model. As shown in Figure 5A, CGK012 and Dex (0.53 mg/kg) significantly reduced TNF-α levels in the BALF of treated mice compared to LPS-only controls. Using the estimated blood volume >of mice (72 mL/kg) 22,23 and the average body weight in this study (27 g), the administered CGK012 doses of 0.06, 0.13, 0.26, and 0.53 mg/kg were equivalent to peripheral fluid concentrations of approximately 2, 5, 10, and 20 μM, respectively. Similarly, Dex at 0.53 mg/kg corresponded to a concentration of 20 μM in peripheral fluid. In lung tissue, CGK012 and Dex both led to a significant reduction in iNOS expression (Fig. 5B), demonstrating the in vivo anti-inflammatory potential of CGK012. Histological assessments further supported the protective effects of CGK012 and Dex, showing considerable alleviation of LPS-induced lung tissue damage (Fig. 5C, D). To elucidate the effects of CGK012 on the Nrf2/HO-1 signaling pathway and its downstream mediators, we analyzed Nrf2 and HO-1 mRNA expression in lung tissues. Additionally, the activities of CAT and SOD, key antioxidants regulated by the Nrf2/HO-1 axis, were assessed. In LPS-challenged mice, significant downregulation of Nrf2 and HO-1 gene expression and reduced activities of CAT and SOD were observed; these were substantially restored by CGK012 treatment (Fig. 5E, F, and Table 1). Further investigation into whether CGK012 modulates the Nrf2/Keap1 pathway, thereby promoting HO-1 expression and attenuating inflammation, revealed that CGK012 treatment (10 μM and 20 μM for 6 h) effectively disrupted Keap1-Nrf2 binding in primary murine osteoblasts (Fig. 5G). These findings indicate that CGK012 exerts anti-inflammatory effects by modulating the Nrf2/Keap1/HO-1 axis, enhancing antioxidant defenses, and reducing inflammation.
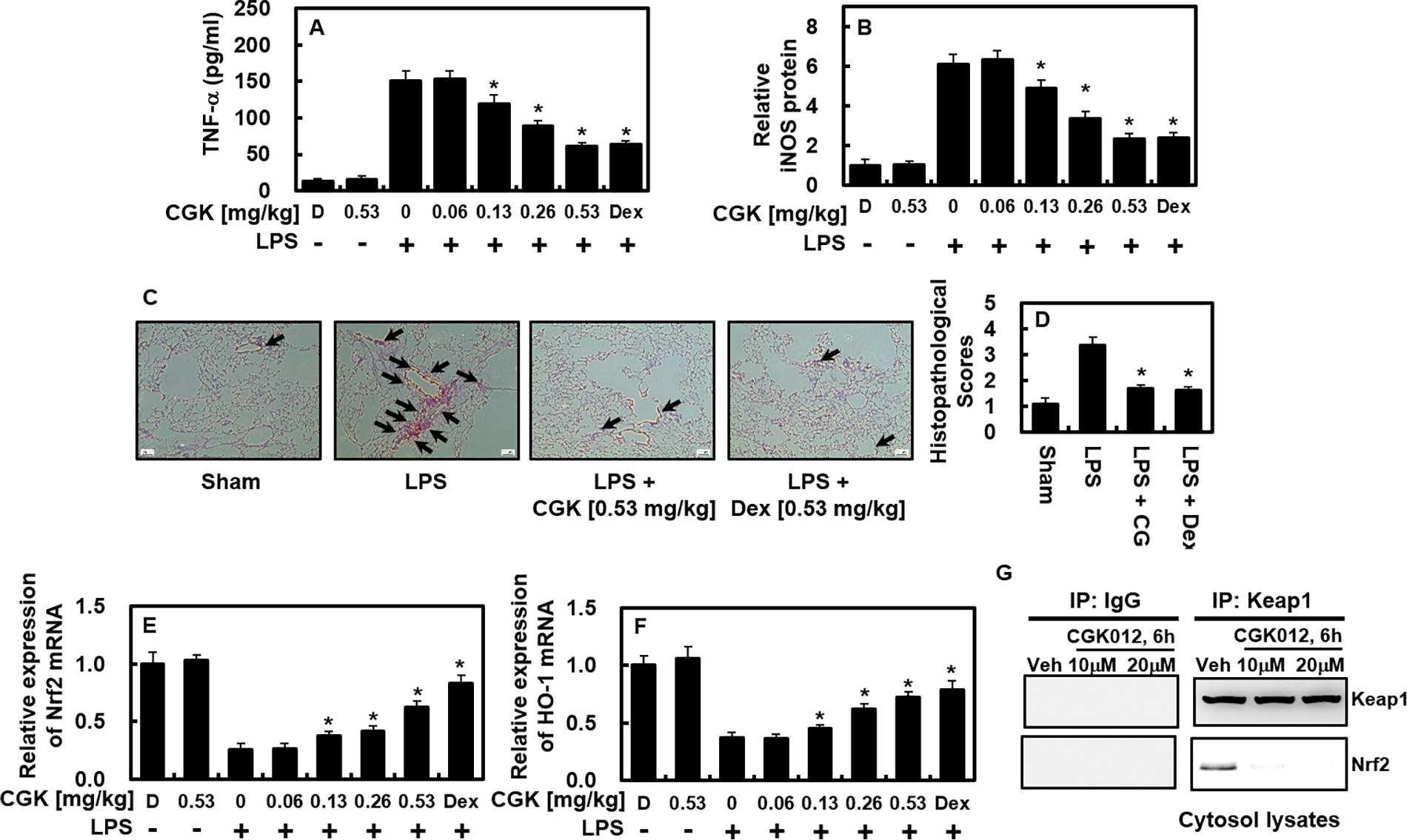
In mice subjected to LPS injection, CGK012 effectively lowered TNF-α and iNOS levels and mitigated lung tissue damage. The experimental protocol consisted of an initial intraperitoneal injection of LPS (15 mg/kg), followed by the administration of CGK012 (at doses ranging from 0.06 to 0.53 mg/kg intravenously) or Dex (0.53 mg/kg) after a 6-h interval. The control group was not subjected to LPS injection. Each experimental group comprised five mice. One day after LPS injection, lung tissue and BALF were collected, and the protein levels of TNF-α
Effects of CGK012 Treatment on the Activities of Antioxidant Enzymes in Lipopolysaccharide-Injected Mice. a
Each value represents the mean ± SD (n = 10).
P < .05 as compared to LPS.
CAT, catalase; DMSO, dimethyl sulfoxide; LPS, lipopolysaccharide; SD, standard deviation; SOD, superoxide dismutase.
DISCUSSION
This study demonstrated that LPS stimulation led to a significant upregulation of inflammatory mediators, including NO, PGE2, TNF-α, and IL-1β, as well as the regulatory enzymes iNOS and COX-2. However, treatment with CGK012 effectively attenuated this increase, indicating its anti-inflammatory and antioxidant effects in both LPS-stimulated cells and in vivo mouse models. Inflammatory responses are characterized by the release of mediators such as NO, PGE2, TNF-α, and IL-1β, alongside the activation of regulatory enzymes iNOS and COX-2. 24,25 Importantly, CGK012 significantly reduced the levels of these inflammatory mediators and enzymes, confirming its potent anti-inflammatory and antioxidant properties. Moreover, CGK012 demonstrated a dose-dependent upregulation of HO-1 expression while concurrently suppressing LPS-induced COX-2/PGE2 and iNOS/NO productions and inhibiting NF-κB activity. NF-κB plays a pivotal role in numerous inflammatory processes, including cell adhesion, proliferation, differentiation, and apoptosis inhibition. 26 It also regulates immune responses by initiating proinflammatory signaling cascades during inflammation. Elevated NO levels, often associated with airway inflammation, contribute to chemokine production, and NF-κB activation is crucial for the expression of COX-2 and iNOS induced by LPS. Our findings suggest that CGK012 suppresses proinflammatory mediator expression (iNOS, COX-2, IL-1β, and NO) by modulating NF-κB activity.
In addition, CGK012’s upregulation of HO-1 may contribute to the downregulation of iNOS and TNF-α expression in BALF from LPS-challenged mice. Both recent and earlier studies have supported the hypothesis that CGK012 enhances HO-1 induction, which inhibits NF-κB activation and oxidative enzyme activity, thus reducing the substrate availability for COX-2 and STAT-1 phosphorylation. Furthermore, CGK012 was shown to increase ARE luciferase reporter activity and to influence the nuclear translocation of Nrf2. Consequently, the anti-inflammatory effects of CGK012 appear to be linked to increased HO-1 expression and reduced iNOS levels, as demonstrated by RNAi-mediated inhibition of HO-1, which reversed the CGK012-induced suppression of NO production and iNOS expression. The present study highlights the ability of CGK012 to enhance HO-1 expression and reduce proinflammatory mediators such as iNOS and TNF-α in LPS-treated HUVECs and lung tissues from LPS-challenged mice. These results underscore the critical role of HO-1 in mitigating inflammatory responses and suggest that TNF-α plays a significant role in this process. Furthermore, our findings indicate that CGK012 regulates inflammation by modulating HO-1 expression, leading to the inhibition of oxidative responses and NF-κB activation. CGK012 promotes HO-1 expression in a dose-dependent manner while concurrently reducing NF-κB activation. Previous research has proposed that activation of the Nrf2 antioxidant pathway may inhibit the transcriptional upregulation of proinflammatory cytokines induced by LPS. 27 Given that CGK012 effectively suppresses proinflammatory cytokines such as TNF-α and IL-1β, it is plausible that its anti-inflammatory effects are mediated, at least in part, through activation of the Nrf2 pathway. To explore this hypothesis, the study examined the expression of Nrf2 and Keap1 in LPS-stimulated cells and observed that CGK012 dose-dependently enhanced the nuclear translocation of Nrf2 while downregulating Keap1 expression. These findings suggest that CGK012 exerts its anti-inflammatory and antioxidant effects, at least in part, through activation of the Nrf2 antioxidant pathway.
The anti-inflammatory properties of CGK012 have been comprehensively investigated in animal models, yielding promising results. This study specifically assessed the impact of CGK012 on TNF-α and iNOS protein levels in a murine model of LPS-induced lung injury. The findings revealed a significant reduction in TNF-α levels within the BALF of CGK012-treated mice, suggesting strong anti-inflammatory activity. In addition, CGK012 substantially decreased iNOS expression in lung tissue, corroborating its efficacy in vivo. Notably, CGK012 also demonstrated protective effects on lung tissue during inflammatory episodes, mitigating the pulmonary damage triggered by LPS exposure. Collectively, these data underscore the potential of CGK012 as a therapeutic intervention for inflammatory diseases, particularly within the respiratory system. Importantly, the dosages of CGK012 administered were extrapolated to peripheral fluid concentrations, providing critical insights that inform future studies and establish a foundation for determining appropriate therapeutic doses for potential human applications. Overall, this research highlights the potent in vivo anti-inflammatory effects of CGK012, especially in the context of lung inflammation, suggesting its potential utility for developing novel therapies aimed at a range of inflammatory disorders. Nevertheless, additional studies are essential to elucidate the molecular mechanisms underlying CGK012’s anti-inflammatory effects and to assess its clinical applicability.
Future research should aim to clarify the molecular mechanisms through which CGK012 upregulates HO-1 and modulates the Nrf2 and NF-κB pathways. Long-term studies evaluating the safety and possible side effects of CGK012, particularly with chronic administration, are crucial. Additionally, optimization of CGK012 dosage and delivery methods across various inflammatory conditions will be essential. Research should also explore CGK012’s efficacy in treating other inflammation-related diseases beyond lung injury, such as arthritis and neuroinflammation. Comparative studies with existing anti-inflammatory therapies would help position CGK012 within the current therapeutic landscape. Finally, human clinical trials assessing the pharmacokinetics, pharmacodynamics, and overall clinical feasibility of CGK012 are vital for advancing its potential as a therapeutic agent.
This study further demonstrated that CGK012 promotes nuclear translocation of Nrf2 (Fig. 4), a pivotal step in activating the Nrf2/Keap1 pathway, as evidenced by increased Nrf2 binding to AREs post-treatment. While direct interaction between CGK012 and Nrf2 or Keap1 was not established, the results strongly indicate that CGK012 influences the Nrf2/Keap1 pathway, potentially through mechanisms involving Nrf2 stabilization or Keap1 inhibition. Additionally, CGK012 was shown to induce HO-1 expression (Fig. 3), a known downstream target of the Nrf2 pathway, indirectly suggesting that CGK012 modulates this axis. The attenuation of inflammation through HO-1 supports the involvement of the Nrf2/Keap1 pathway. Further, CGK012 treatment (10 μM and 20 μM for 6 h) was shown to disrupt the Keap1-Nrf2 association in HUVECs (Fig. 5G), confirming CGK012’s interaction with the pathway.
In conclusion, this study demonstrates that CGK012 effectively elevates HO-1 levels and reduces proinflammatory cytokine production in HUVECs exposed to LPS. Additionally, it decreases iNOS and TNF-α levels in the lung tissues of LPS-exposed mice. These findings underscore the critical role of HO-1 in regulating inflammatory responses and highlight the potential involvement of TNF-α in modulating the HO-1 pathway. Thus, CGK012 represents a promising therapeutic candidate for managing inflammatory conditions, particularly those affecting the respiratory system.
Footnotes
AUTHORS’ CONTRIBUTIONS
J.L. and J.B.H.: Conceptualization, methodology, and investigation. H.J.H. and G.N.: Investigation. G.Y.S. and J.-S.B.: Conceptualization, methodology, supervision, resources, and writing—reviewing and editing.
AUTHOR DISCLOSURE STATEMENT
The authors have no conflicts of interest to declare.
FUNDING INFORMATION
This research was supported by the