
Abstract
Objective:
PD0325901, a selective inhibitor of mitogen-activated protein kinase kinase (MEK), was associated with the occurrence of ocular retinal vein occlusion (RVO) during clinical trials in patients with solid tumors. As previous animal safety studies in rats and dogs did not identify the eye as a target organ of toxicity, this work was conducted to develop a rabbit model of ocular toxicity with PD0325901.
Methods:
Dutch-Belted rabbits were administered a single intravitreal injection of PD0325901 (0.5 or 1 mg/eye) or saline control, and ophthalmic examinations and retinal angiography were conducted over a 2-week period post-dose. In addition, mechanism of ocular toxicity was further explored in rat with microarray analysis.
Results:
PD0325901 treatment produced RVO with retinal vasculature leakage and hemorrhage within 48-h postinjection in Dutch-Belted rabbits. Subsequent retinal detachment and degeneration were also detected on day 8 postinjection. To evaluate the potential mechanism(s) of PD0325901-mediated RVO, male Brown Norway rats were orally administered PD0325901 (45 mg/kg/day) up to 5 days and retinal tissue was collected for gene array analysis. Although PD0325901 did not produce clinical evidence of RVO in rats, retinal gene expression suggested an increased oxidative stress and inflammatory response, endothelium and blood-retinal barrier damage, and prothrombotic effects. Moreover, soluble endothelial protein C receptor (sEPCR), a biomarker for RVO, was elevated in human umbilical vascular endothelial cells (HUVECs) cultured with PD0325901.
Conclusions:
This work has developed a rabbit model of PD0325901-induced RVO that may be used to characterize the cellular and molecular mechanisms of this effect in humans.
Introduction
T
Mitogen-activated extracellular protein kinase kinase (MEK) is a key molecule in the Ras/Raf/MEK/ERK signal transduction pathway. Once phosphorylated by Raf kinase, the activated MEK in turn phosphorylates downstream extracellular-regulated kinase (ERK). ERK further phosphorylates and activates a variety of substrates in the MAPK pathway. There are 2 isoforms of MEK protein, MEK1 and MEK2, which have dual-specific serine/threonine and tyrosine kinase activity and share 80% sequence homology.6 The only known substrates that have been identified for MEK 1/2 are ERKs. Several inhibitors against Raf, MEK, and ERK have been developed and, among them, MEK inhibitors are further along in clinical development. The most notable MEK inhibitors are PD0325901, CI-1040, and AZD6244 (ARRY-142886), which have exhibited antitumor activity in clinical trials.7–9
PD0325901 [N-(2,3-dihydroxy-propoxy)-3,4-difluoro-2-(2- fluoro-4-iodo-phenylamino)-benzamide] is a second generation small molecule inhibitor of MEK 1 and 2 that had been in clinical development for the treatment of solid tumors including breast, colon, non–small cell lung cancer, and melanoma (Pfizer Phase I Clinical Trial Study #4581001). PD0325901 was clinically active against some of these tumor types and the pharmacokinetic (PK) and pharmacodynamic (PD) relationships have been established.10 A small number of cancer patients treated with PD0325901 were diagnosed with retinal vein occlusion (RVO), described by the presence of cotton wool spots, hemorrhages, and vein occlusion.
RVO is considered a clinically significant adverse effect that can lead to vision loss. The pathology of RVO includes poor venous drainage and increased retinal capillary pressure and permeability, which may ultimately lead to retinal ischemia and macular edema. The RVO observed in early clinical trials with PD0325901 was not present in rats or dogs that were administered PD0325901 daily by oral gavage for up to 3 months. The lack of retinal vasculature or ocular toxicity in normal healthy animals is not entirely unexpected, as the development of RVO in humans has been associated with hypertensive, atherosclerotic, inflammatory, and thrombophilic conditions.11 This study was designed to develop an animal model of RVO to investigate the potential mechanism(s) of this toxicity. Because PD0325901-induced RVO in human patients required at least 3 months of treatment, this study investigated whether direct injection (intravitreal injection) in rabbit eyes would manifest ocular toxicity within a 2- to 4-week period. Rabbits were selected because the rabbit eye is anatomically similar to that of humans with regard to size and structure and the conduct of ophthalmic exam, and fluorescein angiography for retinal vessel examination is well precedented in this species. Intravitreal injection was utilized to ensure that exposure of PD0325901 to ocular tissues was sufficient to produce ocular toxicity, if present. Moreover, studies were also conducted in rats by oral gavage, the clinical route of administration, to determine whether gene expression signatures in the retina may provide evidence of ocular toxicity in the absence of pathologic RVO.
Methods
Animals
Pigmented Dutch-Belted rabbits (female, 2–3 months old, and 2–2.5 kg) were obtained from Myrtle’s Rabbitry, Inc. (Thompson Station, TN). Pigmented Brown Norway rats (male, 6–8 weeks old) were purchased from Charles River Laboratory (Kingston, NY). Animals were given free access to food (rabbits: Certified High Fiber diet #5325, Newco Inc., Rancho Cucamonga, CA; rats: Lab Diet #5001, Newco Inc.) and water, and had standard housing conditions (12 h light/dark cycle). The study was conducted in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All procedures involving laboratory animals were reviewed and approved by the Institutional Animal Care and Use Committee associated with the facility in which the laboratory animals were housed.
Cell cytotoxicity in ARPE-19 cells
A general assessment of the cell-cytoxicity profile of PD0325901 was determined to help guide dose selection for intravitreal toxicity studies in rabbits. Human retinal pigment epithelial cells (ARPE-19, ATCC biological resource center, Manassas, VA) were seeded at a concentration of 10,000 cells/well on clear 96-well plates and exposed to PD0325901 at various concentrations for 24 h. Lactate dehydrogenase (LDH) was measured as a marker of cellular toxicity using the CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega, Madison, WI) as per manufacturer’s instructions.
Animal treatment
Rabbits were used to assess the ocular toxicity of PD0325901 following intravitreal injection into the eye. Briefly, animals were anesthetized with xylazine (3 mg/kg) and ketamine (10 mg/kg) by intravenous injection via the marginal ear vein, the eyes were dilated with 1–2 drops of AK-Dilate (phenylephrine) and AK-Pentolate (cyclopentolate) and the eyes underwent sterile preparation for intravitreal injection as previously described.12 Following sterile cleansing of the eye, a 27 gauge needle was inserted into the mid-vitreous ∼3 mm posterior to the limbus in the superior/temporal quadrant. Animals (2–5/group) received PD0325901 (0.5 or 1 mg/eye), CI-1040 (1 or 2 mg/eye), or vehicle (normal saline with 2% polyvinyl alcohol, pH was adjusted to 5.5–6, and osmolarity at 300 mOsm/kg) in both eyes at a dose volume of 0.1 mL/eye, and were observed for 2 weeks. PD0325901 and CI-1040 were formulated as homogeneous suspensions. CI-1040, a MEK inhibitor, was used as a comparator for PD0325901.
To determine whether PD0325901 can produce molecular changes in the eye, an ocular gene expression study was conducted in rats. Rats (n = 3/group) received PD0325901 (45 mg/kg/day) or vehicle (0.5% methylcelluose) via oral gavage for 3 or 5 days. On the day of termination, a necropsy was performed and the retina was collected for gene expression analysis. At several time points throughout the study ophthalmic examination and clinical pathology parameters were collected. The dose level of 45 mg/kg/day was selected to achieve 70% of the maximum tolerated dose for 1 week of dosing.
Ophthalmic examination
Ophthalmic examinations (slit-lamp ophthalmoscope; Kowa Optimed Inc., Torrance, CA) were conducted in rabbits and rats as previously described12 pre-dose and at several time points post-dose. Briefly, rabbits were subjected to light anesthesia (1.5 mg/kg xylazine/5 mg/kg ketamine) and fundus microscope or angiogram photographs were taken pre- and post-dose (day 1, 2, 8, and 15) using a Carl Zeiss Meditec’s FF450 plus fundus camera (Carl Zeiss, Jena, Germany). For angiography, sodium fluorescein (100 µL of 10% AK-FLUO) was administered by intravenous injection and a series of angiogram photographs were taken with blue light every 30 s during a 5-min interval. For rats, the fundus and angiogram examination were performed on days 1 (pre-dose), 2, and 5 with addition of a 90 diopter lens to the camera.
Clinical pathology evaluation
In the rat study, blood (5 mL) was collected via the inferior vena cava at termination for assessment of hematology, clinical chemistry, and coagulation parameters (prothrombin time (PT), activated partial thromboplastin time (APTT), and fibrinogen). Hemotological parameters were measured using Advia 120 Automated Hematology Analyzer (Roche Diagnostics Inc., Indianapolis, IN). Serum clinical chemistry was analyzed using Advia 1200 Chemistry Analyzer (Roche Diagnostics Inc., Indianapolis, IN). Coagulation parameters were assessed with Electra 1400C Coagulation System (Beckman Coulter Inc., Fullerton, CA).
Retinal gene expression analysis in rats
At the 3- and 5-day termination time points, retinal tissue from vehicle- or PD0325901-treated rats was collected and homogenized in Qiazol (Qiagen, Valencia, CA), and RNA was extracted using the RNeasy kit per manufacturer’s instructions (Qiagen, Valencia, CA). RNA (100–200 ng) underwent 2-step cDNA amplification and target preparation (Affymetrix) for hybridization on the GeneChip® Rat Genome 230 2.0 Array. GeneSpring software (Agilent, Sunnyvale, CA) was used for quality control assessment, filtering, and statistical analysis of the data. Following RNA preprocessing steps, initial filtering was done to include only probe sets with signal intensity values between 20th and 100th percentile in one of the 8 samples for further analysis. One-way ANOVA was performed to identify differentially expressed genes (P < 0.05 with Benjamini-Hochberg multiple testing correction). Of these differentially expressed genes, a threshold of 1.5-fold change relative to the control group was applied and analyzed for biological pathways and functions using Ingenuity Pathways Analysis (Ingenuity, Redwood City, CA).
Histopathology
Rabbit and rat eyes were collected for microscopic evaluation at termination. Briefly, rabbit eyes were enucleated and placed in a 6% glutaraldehyde fixative solution and were marked with 2 sutures: one indicating the injection site and the other indicating the superior portion of the globe. The surrounding eyelid and conjunctival tissue were placed in 10% neutral-buffered formalin for histological analysis. Sectioning was performed on a horizontal plane from anterior to posterior. One section included the intravitreal injection site and one included the optic disc. Rat eyes were collected in Davidson’s Blue solution. Following fixation, rabbit and rat eyes were embedded in paraffin blocks, sectioned (5 μm) onto glass slides, and stained with hematoxylin and eosin (H&E) for microscopic evaluation.
sEPCR ELISA assay
To determine the effect of PD0325901 on the level of soluble endothelial protein C receptor (sEPCR), human umbilical vascular endothelial cells (HUVECs; ATCC biological resource center, Manassas, VA) were exposed to PD0325901 or CI-1040 (40 or 100 nM for both compounds) or vehicle for 24 h and the supernatant was collected and assayed by an ELISA kit (Diagnostica Stago, Asnieres, France).
Statistical analysis
Differences between treatment groups were determined using Dunnett’s t-tests. The differential expression of genes was analyzed by one-way ANOVA (P-value <0.05 was considered to be statistically significant).
Results
PD0325901-induced retinal vein occlusion in rabbits
To guide dose selection for intravitreal injection, the cytotoxicity profile of PD0325901 was determined in vitro in human retinal pigment epithelial cells. A dose-dependent increase in cytotoxicity was observed with PD0325901 treatment as measured by LDH release into media (Fig. 1). PD0325901 produced measurable cytotoxicity at 700 µg/mL, while concentrations below this were minimally cytotoxic. Thus, a concentration of 700 µg/mL in the eye was targeted as a level that may serve as a threshold dose for cytotoxicity. When considering the volume of the rabbit vitreous (1.4 mL), a dose of 1 mg/eye was extrapolated to be a potentially toxic dose and a lower dose of 0.5 mg/eye (equivalent to 350 µg/mL) was selected as a subtoxic dose.
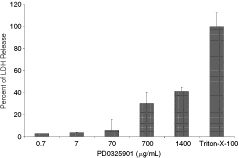
Human retinal pigment epithelium cells (ARPE-19) were seeded at a concentration of 10,000 cells/well on clear 96-well plates. Cells were exposed to PD0325901 (0.7, 7, 70, 700, 1,400 µg/mL) or Triton X-100 (positive control for maximum LDH release) for 24 h and lactate dehydrogenase (LDH) was measured. Data are presented graphically as mean ± SD.
Immediately following intravitreal injection of PD0325901, a drug depot in the vitreous body was observed by fundus examination (Fig. 2A) with both the 0.5- and 1-mg/eye doses. Within 24 h of treatment, the 1-mg/eye dose produced retinal hemorrhages in multiple areas throughout the eye as observed by ophthalmic examination. One week postinjection, the retinal findings progressed to retinal detachment, edema, and retinal vasculature attenuation in this dose group (Fig. 2B and 2C). Ophthalmic examinations did not identify abnormal findings in eyes treated at the 0.5 mg/eye dose. Fluorescein angiography was utilized as a more sensitive method to assess potential changes in retinal vasculature following PD0325901 injection. The fundus angiograms showed obvious vascular leakage, sodium fluorescein obstruction, capillary loss, and abnormal kinetic blood flow (Figs. 2E, 2F, and 3B) at the 1 mg/eye dose. Retinal vessel leakage was also observed with the 0.5 mg/eye dose of PD0325901 in the absence of retinal vasculature occlusion. Fundus images revealed major retinal branch narrowness, stasis, and alterations in retinal vascular geometry (angles or vessel tortuosity) in all eyes treated with 1 mg/eye of PD0325901 (Fig. 3). By contrast, CI-1040 or vehicle treatment did not produce any signs of ocular toxicity.

PD0325901 was administered by intravitreal injection to rabbits; fundus photographs (

PD0325901 was administered by intravitreal injection to rabbits; fundus photograph and angiogram were collected at day 15 post-dose. Fundus image of major retinal branch stasis and abnormal appearance with adjacent vasculature (
Microscopic evaluation of the eye following intravitreal injection of PD0325901 identified moderate and diffuse retinal detachment, mild-to-moderate retinal degeneration and vacuolation, optic nerve vacuolation, and plasma cell infiltrates in the nuclear la yer in only the 1-mg/eye dose group. The degeneration was characterized by multifocal-to-diffuse disorganization of the retinal layers with loss of nuclei from the outer and inner nuclear layers, vacuolation of the inner nerve fiber, inner nuclear layer and outer nuclear layer, and collapse of the photoreceptor layer (Fig. 4). Abnormal ocular findings were not observed with CI-1040 treatment.
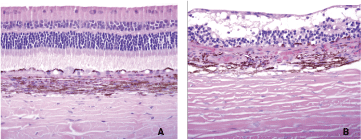
PD0325901 was administered by intravitreal injection to rabbits; eyes were collected at day 15 post-dose. Control-treated eye (
PD0325901-induced systemic and retinal changes in rats
Rats were orally treated with PD0325901 to determine the potential for ocular toxicity by assessing changes in retinal vasculature and retinal gene expression analysis. The 45 mg/kg/day dose of PD0325901 achieved the targeted 70% MTD given the progressive body weight loss of up to 14% compared to control animals by the end of the 5-day treatment period. Ophthalmic examinations or fundus fluorescein angiography did not reveal any clinical evidence of posterior ocular toxicity in PD0325901-treated rats, except for cornea dry spots in anterior segment. However, PD0325901 produced a number of changes in retinal gene expression in a time-dependent manner. A subset of genes statistically significantly altered by 1.5-fold of control is provided in Table 1. Among the significantly altered genes on day 3, oxidative stress response gene αB-crystalline (CRYAB) was the most upregulated (+1.4 log2 ratio to the control); vascular cell adhesion molecule 1 (VCAM1) (+0.7) and acute phase response protein lipocalin 2 (LCN2) (+0.6) were also upregulated; the only downregulated gene was a Müller cell–specific water channel, aquaporin 4 (Aqp4) (–1.1 log2 ratio to the controls). Among the significantly perturbed genes on day 5 were those involved in oxidative stress and acute phase responses, cytokine signaling and coagulation cascade, leukocyte extravasation, and water/osmolyte transportation. The most notable genes that were altered included CRYAB (5.2-fold), heat shock protein 1 (HSPB1) (+4.2), lipocalin 2 (LCN2) (+2.0), coagulation factor VIII (F8) (+2.0), von Willebrand factor (VWF) (+1.8), Rho GTPase activation protein 1 (ARHGAP1) (+1.5), TIMP metallopeptidase inhibitors 2 and 3 (TIMP2 and 3) (+1.6 and +2.2), plasminogen activator/urokinase (PLAU) (+1.8), myosin light chain kinase (MLCK) (+1.6), endothelial protein C receptor (EPCR) (+1.0), aquaporin 4 (AQP4) (–1.0), potassium voltage-gated channel, shaker-related subfamily, members 4 and 5, beta member 1 (KCNA4 and 5, KCNAB1) (–2.5, –1.8, –1.6, –1.3), potassium channel, subfamily K, members 1 and 2 (KCNK1 and 2) (–0.9, –1.8).
E
Values are expressed as log2 ratios of gene expression signals in treated versus control samples. Genes are sorted alphabetically within each sub-section; those induced at 1.5-fold or greater are represented in bold, and those repressed at 1.5-fold or greater are represented in italics. Some genes appear in more than one sub-section due to their diverse roles.
Compared with the vehicle control, rats that received PD0325901 had an 8% reduction in RBC count, hemoglobin, and hematocrit, and a 54% decrease in reticulocyte count by day 3 (Table 2) suggesting a mild anemia in the animals and a suppressive effect of PD0325901 on erythropoiesis, which is consistent with the clinical trial results with both PD0325901 and CI-1040.13 The blood coagulation analysis revealed that the 3-day PD0325901 treatment significantly increased plasma fibrinogen (2.2-fold relative to the control) and slightly decreased PT and APTT (Table 2).
E
∗P < 0.05,
∗∗P < 0.01, compared to the control; n = 3. Data are expressed as mean ± SD.
Abbreviations: APTT, activated partial thromboplastin time; HCT, hematocrit; HGB, hemoglobin; PLT, platelet; PT, prothrombin time; RBC, red blood cell count; Retic, reticulocyte count.
sEPCR level in cell culture
A potential biomarker for RVO, sEPCR, was assessed in HUVECs following exposure to PD0325901 or CI-1040. PD0325901 produced a greater release of sEPCR from HUVECs into media than CI-1040 (100 vs. 5–20 ng/mL; Fig. 5). The amount of sEPCR released with PD0325901 was 24-fold greater than that with CI-1040 at the 40-nM exposure level, and 5 times greater at 100-nM dose level. These results suggest that PD0325901 has a substantial inductive effect on sEPCR secretion and the effect is much stronger than that observed with CI-1040.
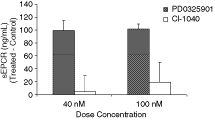
Human umbilical vascular endothelial cells (HUVECs) were treated for 24 h with PD0325901 or CI-1040 at a concentration of 40 and 100 nM. The induction of soluble endothelial protein C receptor (sEPCR) was significantly higher in PD0325901 treatment over CI-1040 at both dose concentrations (P < 0.001). The concentrations shown are the absolute values after subtraction of the vehicle control values. Data are presented graphically as mean ± SD.
Discussion
The oculovisual system has a potentially high degree of susceptibility to toxic substances. The ocular adverse side effects caused by systemic drug administration cover a broad spectrum of disorders from the anterior to posterior chambers of the eye that include inflammatory reactions of the conjunctiva and cornea, blurred vision, ocular pain, photophobia, glaucoma, cataract, retinopathy, optic neuropathy, and even blindness.14 Ocular toxicity induced by systemic anticancer chemotherapy is not uncommon and may be increasing because of the greater number of chemotherapeutic agents and longer patient survival.14 A number of anticancer drugs and experimental compounds that have been reported to cause retinopathy include interferon,15 tamoxifen and toremifene,16 cisplatin,17 , 18 mitotane (an agent that alters steroid peripheral metabolism),19 , 20 and recently, PD0325901. In general, the mechanisms of ocular toxicity elicited by these drugs are not well understood.
PD0325901 is a second generation MEK inhibitor that has shown some promise as a potential antitumor agent in early clinical trials.21 In the phase I dose escalation clinical trial, PD0325901 was associated with an increased incidence of RVO (Pfizer Phase I Clinical Trial Study #4581001). RVO was observed in patients who received higher doses (10–30 mg BID) over a relatively shorter period (12 weeks) or at a lower dose over a longer period (36 weeks) of treatment. This suggests that PD0325901-induced RVO is both dose and/or duration dependent. This is the first report of a MEK inhibitor to be associated with ocular toxicities such as RVO. Considering the interest of developing MEK inhibitors as novel anticancer agents, it is important to study the mechanism of this adverse ocular side effect.
The objective of this work was to establish animal models of PD0325901-induced ocular toxicity to better study the mechanisms of this effect. We demonstrated that direct administration of PD0325901 in rabbit eyes produced effects of RVO similar to those observed in cancer patients. For example, retinal vascular occlusion and vascular hemorrhage and leakage were observed in the rabbit model following PD0325901 treatment that supports the use of this model to further understand the mechanism(s) of drug-induced RVO and the pathogenesis of RVO, as a whole.
To investigate the role of MEK inhibition in the development of PD0325901-induced RVO, CI-1040, a first generation MEK inhibitor and structural analogue of PD0325901, was also evaluated as a comparative agent. In contrast to the findings with PD0325901, CI-1040 did not cause any retinal abnormalities following direct injection into rabbit eyes, and correspondingly, induced much less sEPCR release in cell culture than PD0325901. In clinical trials, CI-1040 had a well-tolerated safety profile without any evidence of retinal vasculature toxicity. However, CI-1040 is much less potent than PD0325901, as their IC50 against MEK are 2 μM and <1 nM, respectively.21 Thus, the insufficient clinical efficacy observed with CI-1040 and the relatively high dose (800 mg BID) required in the phase II testing, contributed by its poor bioavailability and metabolic instability,22 resulted in the discontinuation of its clinical development in oncology. Notably, another MEK inhibitor is also in clinical testing for oncology, AZD6244 (ARRY-142886). Compared to PD0325901, AZD6244 (IC50 10 nM) is 10 times less potent, has a relatively poor bioavailability, and lower pERK inhibition in tumor biopsies. Its clinical dosing regimen was 50–300 mg BID with a duration of 16–40 weeks.21 Although no incidence of RVO has been reported with AZD6244, blurred vision occured in about 12% of patients in the phase I trial.23 Therefore, potency of MEK inhibition tracks with ocular toxicity in humans. The compound potency, bioavailability, and dosing duration likely play key differentiating factors for RVO induction, which may explain the lack of effects observed with CI-1040 in our rabbit model. Testing the AZD6244 compound in rabbits would be fruitful in determining the role of MEK inhibition in RVO. A more direct link between RVO and MEK inhibition could be ascertained using antisense oligonucleotides or siRNA to knockdown MEK expression in the eye. The potential roles of MEK1 and MEK2 in the ocular toxicity may be differentiated using this approach because their expression profiles and biological functions do not completely overlap.24–26
Further evidence to support the link of RVO to MEK inhibition may be found in studying interferon-alpha-2a (IFNα2a)-induced RVO. RVO has been widely reported in cancer or hepatitis C patients undergoing IFNα2a therapy.27 , 28 The typical retinopathy is described to include cotton wool spots, retinal hemorrhages, macular or papillary edema, and retinal or even choroidal vascular occlusion, which occur by 8–12 weeks of treatment. Although the number of clinical reports of retinopathy related to interferon therapy is increasing, the mechanisms of ocular toxicity remain unsolved. However, a few studies show a close relationship between IFNα2a and MEK/MAPK signaling pathways. Romerio and colleagues (2000) reported that IFNα2a inhibited the activities of MEK and ERK through a Ras/Raf-independent mechanism and Romerio and Zella and Battcock and colleagues reported that the MEK/MAPK pathway negatively regulated the interferon response and inhibition of the pathway would enhance the interferon effect.29–31 These reports strengthen the speculation that RVO induced by either interferon or PD0325901 may share a similar molecular mechanism.
Oral administration of PD0325901 did not lead to ocular toxicity based on previous toxicity studies that were up to 13 weeks of duration in rats and dogs (unpublished data). Because changes at the molecular level were more sensitive, microarray gene expression profiling was performed on retinal tissue following oral dosing in rats. Indeed, the gene expression analysis of the retina isolated from the PD0325901-treated rats identified changes in oxidative stress, acute phase and inflammatory response, blood-retinal barrier (BRB) breakdown, leukostasis, and coagulation cascade activation, which may all contribute to the onset of RVO retinopathy (Table 1; Fig. 6).

Proposed mechanism for PD0325901-induced retinal vein occlusion and edema. Induction of antioxidant genes and antigen presentation gene β-2-microglobulin suggests the occurrence of oxidative stress in the retina following PD0325901 treatment. Oxidative stress is closely associated with inflammatory responses, which may lead to impaired blood-retinal barrier (BRB) integrity and leukocyte extravasation. Downstream consequences include retina edema, hemorrhaging, and subsequently the activation of the coagulation cascade, all of which may contribute to the onset of retinal vein occlusion (RVO) and retinal edema.
A potential impact of oxidative stress and inflammation on endothelial cells is an increase in vascular permeability, which would impair the integrity of the BRB. Vascular endothelial cells, together with Müller cells and retinal pigment epithelium, make up the BRB.32 In this study, the expression of myosin light chain kinase (MLCK) was significantly increased, likely as a result of TNFR/NF-κB signaling. Phosphorylation of myosin light chain by MLCK could lead to actin-mediated endothelial cell contraction and consequently increase permeability of endothelial junction barrier.33 Activation of phosphatidylinositol 3-kinase (PI3K), whose expression was induced on both day 3 and day 5, has also been shown to increase vascular permeability.34–36 In addition, matrix metalloproteinase 14 (MMP14) was induced on day 5. MMP14 activity can lead to cleavage of vascular endothelial (VE)-cadherin,37 which forms part of the adherens junction important for vascular endothelium integrity. On the other hand, our data set also uncovered induction of genes important for maintaining the vascular endothelial barrier function, a potential as feedback mechanism to combat permeability increases, such as the repression of RhoA and the induction of Rac1 to facilitate reannealing of adherens junctions,33 the induction of claudin 11 and junctional adhesion molecule (JAM) 2 may restore tight junction integrity,38 and the induction of pigment epithelium–derived factor (PEDF, serpinf1) may repress the expression of VEGF and, therefore, decrease vascular permeability.32
Müller cells also play a vital role in the maintenance of BRB integrity.32 Under physiological conditions, Müller cells carry out transcellular water transport from the retinal interstitial space into the blood, thus preventing excess fluid buildup within the retina. The transcellular water transport is osmotically coupled to the transport of potassium ions. Under conditions of oxidative stress and inflammation, Müller cells have been shown to contribute to retinal edema through a disturbed intracellular fluid transport. In this study, the repression of the Müller cell water channels (Aqp4 and 11) coupled with the repression of potassium channels signals impaired transcellular fluid transport, which may lead to cell injury and edema. Taken together, microarray data suggest a molecular event of increased retinal endothelial cell permeability, impaired BRB function, and fluid imbalance.
Interestingly, diabetic or hypertensive rats may be more sensitive to PD0325901-mediated RVO, as the vasculature in these disease conditions is often compromised. As we have learned, PD0325901 can trigger a series of pathogenic molecular changes in the normal retina. When retinas are compromised by pre-existing pathological conditions, especially of vascular or diabetic diseases, they may be more susceptible to the effects of MEK inhibition, which could explain the variable incidence of RVO in the clinical trial results.
Hematological dysfunction, such as increased plasma fibrinogen and disruption of the thrombosis-fibrinolysis balance, has been implicated in the development of RVO.39 Fibrinogen is a key player in the coagulation cascade that induces platelet aggregation and is the substrate for fibrin formation. Increased fibrinogen has been associated with RVO in several clinical reports.40–42 Further, the thrombin inhibitor argatroban has been shown to reduce leukocyte-endothelial cell interactions and retinal vascular leakage.43 Following oral administration of PD0325901 in the rat, plasma fibrinogen levels were significantly increased and correlated with increased gene expression of fibrinogen, coagulation factor VIII, and von Willebarnd factor in the retinas. In addition to its role in the coagulation cascade, fibrinogen is also an acute-phase reactant that becomes elevated with tissue inflammation or tissue destruction, and may be a predictor for circulatory disease. Therefore, the significant increase in fibrinogen could indicate tissue inflammation, prothrombotic state, and vascular abnormalities in PD0325901-treated animals.
sEPCR, another hemostasis and inflammation regulator, has been proposed to be a risk factor for RVO.44 The endothelial cell protein C receptor (EPCR, also known as CCD41 or CD201) is mainly expressed on the endothelial cells of vessels and functions as an anticoagulant factor in the protein C anticoagulant pathway by binding protein C and enhancing its activation by the thrombin-thrombomodulin complex. The soluble form of EPCR (sEPCR) has recently been detected in normal human plasma and in contrast to membrane-bound EPCR, plays a prothrombotic role by inhibiting protein C activation and competition with EPCR.45 , 46 sEPCR also binds to factor VIIa and inhibits its coagulant activity.47 In this study, the level of sEPCR protein was significantly higher in the media of PD0325901-treated HUVEC cultures than that in the media of CI-1040 treated cultures. Consistent with this finding, EPCR gene expression was induced in the retinas of PD0325901-treated rats. Considering that CI-1040 was not associated with RVO in human or animals in contrast to PD0325901, the increased sEPCR induced by PD0325901 suggests a meaningful association between RVO and sEPCR and supports its potential use as a biomarker for RVO.
In conclusion, intravitreal injection of PD0325901 in rabbits reproduced the ocular toxicity observed in humans. The retinopathy produced in the PD0325901-treated rabbit eyes was similar to retinal vascular occlusion, hemorrhage, and leakage observed in cancer patients treated with the inhibitor. Intravitreal delivery with intensive ophthalmic examinations including fundus microscopy and angiography may represent a viable screening approach for potential ocular toxicity caused by therapeutic compounds. Investigation into the molecular events in clinically and histologically normal retinas of PD0325901-treated rats suggested oxidative stress and inflammatory responses, endothelial cell injury, impaired blood-retina barrier, dysregulated fluid transport, and prothrombotic activities. Based on this study, it would be reasonable to run de-risking studies using this rabbit RVO model for next generation MEK inhibitors to minimize the potential for later stage attrition. This strategy could also be extended to other drug targets within the MAPK cascade, such as RAS, RAF, and ERK during candidate selection.
Footnotes
Acknowledgements
The authors thank Leigh Ann Burns-Naas, Alejandro Ricart, and Greg Stevens for discussion and critical review.