
Abstract
Abstract
Purpose:
Recently, cell cycle reentry has been reported in the experimental brain ischemia model and Alzheimer's disease brain. Cyclin-dependent kinases (CDKs) are known to be key molecules that regulate the cell cycle. The aims of the present study were to determine the change in expression of the proteins involved in regulation of G1 phase, and to test the effect of small molecule CDK inhibitors on neuronal injury in the ischemic-reperfused rat retina.
Methods:
Male Sprague-Dawley rats were anesthetized and subjected to 60 min of retinal ischemia by raising intraocular pressure to 130 mmHg. One day after this procedure, the retinal tissues were homogenized and immunoblot analyses were done to check the expression levels of cell cycle regulatory proteins. Additional groups of rats received 3 mg/kg roscovitine, a CDK inhibitor, and 1 mg/kg CDK4 inhibitor intravenously (i.v.) 15 min before ischemia and underwent 60 min of ischemia. On the 7th day of the retinal ischemia-reperfusion, eyes were subjected to morphometry.
Results:
Immunoblot analysis revealed that ischemia-reperfusion significantly increased expression levels of cyclin D1 and CDK4, and decreased those of p16INK4 and p27KIP1. Treatment with a CDK4 inhibitor (1 mg/kg, i.v.) 15 min before ischemia significantly reduced death of the retinal ganglion cells.
Conclusion:
Upregulation of cell cycle regulatory proteins and activation of nonmitotic CDK5 are shown to be associated with neuronal cell death. Although the CDK4 inhibitor used in the present study is known to reduce the activity of mitotic CDK4, there is no information about its blocking effect on other CDKs, such as nonmitotic CDK5. The results in the present study suggest that abnormal progression of the cell cycle and/or activation of nonmitotic CDK are involved in the neuronal cell death induced by retinal ischemia-reperfusion. Furthermore, as shown with a CDK4 inhibitor, agents that alter the activity of CDK may be good candidates for inhibitors of neuronal cell death induced by ischemia-reperfusion injury.
Introduction
Neurons are differentiated terminally and unable to reenter the cell cycle in general. However, evidence reported recently has shown upregulation of cell cycle regulatory proteins in degenerating neurons. 5 Upregulation of cyclin-dependent kinase (CDK) 4, CDK 5, cyclin A, and cyclin D and downregulation of p16INK4 have been reported in in vivo stroke models6,7 and pathologically affected or vulnerable neurons in Alzheimer's disease. 8 These findings suggest that certain cell cycle regulators play a critical role in the neurodegenerative process. Drugs that block the progression of cell cycle reduced cell death in not only PC12 cells but also sympathetic neurons. 9 Flavopiridol, a general CDK inhibitor, is reported to be protective in both focal 10 and global 11 brain ischemia. These lines of evidence suggest that mature neurons still keep the function of certain elements of the cell cycle, and that they could reactivate the replication mechanism under certain stresses, and that events that induce cell cycle reentry in the mature neurons are lethal rather than mitogenic.
Both Alzheimer's disease and glaucoma are chronic neurodegenerative diseases. Recent reports suggest that there are some similarities between Alzheimer's disease and glaucoma, such as activation of caspase and abnormal processing of amyloid precursor protein. 12 Bayer et al. 13 reported that patients with Alzheimer's disease and Parkinson's disease had a higher risk of glaucoma. Donepezil, a therapeutic drug for Alzheimer's disease, is recently reported to have a protective effect against RGC death induced by ischemia-reperfusion, 14 glutamate, 15 and axotomy. 16 The drug is reported to reduce the number of the cells in the S-G2/M phases and parallelly increased the G0/G1 population in the SH-SY5Y cells. 16 In addition, expression of 2 cyclins of the G1/S and G2/M transitions, cyclin E and cyclin B, was significantly reduced, whereas expression of the cell cycle inhibitor p21 increased after exposure to the drug in the cells. 16 Taken together, the observations stated above suggest that abnormal progression of the cell cycle may be involved also in the neuronal degeneration induced by retinal ischemia-reperfusion.
Nonmitotic CDK5 is a key regulator of neuronal death and survival. 7 The activity of CDK5 in the brain is triggered by p39 and p35, which are its binding partners. 17 Lee et al. have shown that glutamate-induced excitotoxicity on neuronal culture produced a cleavage of p35 in p25 by calpain. 18 The p25 fragment induces hyperactivation of CDK5 and translocation of the p25/CDK5 complex to the cytoplasm. The complex hyperphosphorylates a number of substrates, and leads to neuronal death. 19 Lefèvre et al. suggested that axotomy-induced death of RGCs involves the activation of CDK5 in the chicken embryos. 20 Menn et al. recently reported that systemic (S)-roscovitine provided neuroprotection with inhibition of CDK5 activity increase in that rat forebrain stroke model. 21
Although previous reports suggest that mitotic CDKs and nonmitotic CDK5 may be good therapeutic targets for ischemia-reperfusion injury, it is still unclear whether the expression levels of the proteins involved in regulation of G1 phase were changed in the ischemic-reperfused rat retina, and whether small molecule CDK inhibitors, some of which are known to inhibit both mitotic CDKs and CDK5, are protective against ischemia-reperfusion injury in the retina. The aim of the present study was to determine the changes in expression of the proteins involved in regulation of G1 phase, and the effects of small molecule CDK inhibitors on neuronal cell injury in the ischemic-reperfused rat retina.
Methods
Animals
In the present study, experimental procedures conformed to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research and the Guiding Principles for the Care and Use of Laboratory Animals, approved by the Japanese Pharmacological Society. Male Sprague-Dawley rats weighing 230–300 g (Charles River Japan, Kanagawa, Japan) were anesthetized with pentobarbital sodium (50 mg/kg intraperitoneally; Nembutal® injection; Abbott Laboratories, North Chicago, IL), and the rectal temperature of animals was maintained at 37°C during experiment using heating pad and heating lamp. The femoral veins were cannulated for administration of the drug if necessary.
Induction of retinal ischemia
Induction of retinal ischemia was performed as previously described.14,22–25 Briefly, the anterior chamber of the 1 eye was cannulated with a 27-gauge needle connected to a bottle filled with saline. Retinal ischemia was induced by raising intraocular pressure to 130 mmHg by lifting the bottle for 60 min. The opposite eye of each animal served as a nonischemic control.
Immunoblotting
After sacrifice, retinas from enucleated eyes were dissected using a dissection microscope. The retinas were homogenized with cell lysis buffer solution comprised of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/mL aprotinin, and 1 mM leupeptin, using a homogenizer (Miccra D-8; ART Labortechnik, Müllheim, Germany). The homogenized samples were then centrifuged at 16,000 g for 30 min at 4°C. The supernatant was used for immunoblotting. Total protein in samples was determined according to Lowry et al. 26 Aliquots of each fraction containing equal quantities of protein were separated on 12% polyacrylamide gels. The separated proteins were transferred electrophoretically to PVDF membranes (Biorad, Hercules, CA) and blocked by 10% skim milk in phosphate-buffered saline (PBS)-Tween 20, comprised of 137 mM NaCl, 2.6 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, 0.05% Tween 20, and probed with anti-p16INK4 antibody (1:100; Santa Cruz, Santa Cruz, CA), anti-p27KIP1 antibody (1:100; Santa Cruz), anti-CDK4 antibody (1:100; Santa Cruz), or anti-cyclin D1 antibody (1:100; Nichirei Bioscience, Tokyo, Japan). Horseradish peroxidase (HRP)-labeled anti-mouse immunoglobulin G (IgG) antibody or HRP-labeled anti-rabbit IgG antibody was used for the secondary antibody. Western Lightning chemiluminescence reagent Plus (Perkin-Elmer, Norwalk, CT) was used to observe immunoreactions on Hyperfilm ECL (Amersham, Buckinghamshire, UK). The band density was measured with ImageJ software (National Institute of Health, Bethesda, MD).
Histological evaluation
Histological evaluation was performed as previously described.14,22–25,27 In brief, 5-μm horizontal sections through the optic nerve head of the eye were stained with hematoxylin and eosin, and subject to morphometry. The total number of the cells in the retinal ganglion cell layer (GCL) was counted for a length of 1 mm on either side of the optic nerve head beginning approximately 1 mm from the center of the optic nerve head in 4 independent sections using a light microscope (Optiphot-2; Nicon, Tokyo, Japan). Measurement of the thickness of the IPL, the inner nuclear layer (INL), and the outer nuclear layer (ONL) was also performed to quantify the degree of cell loss induced by retinal ischemia-reperfusion. These parameters of each eye subjected to ischemia were normalized with those of the corresponding intact opposite eyes and are presented as percentages.
To examine whether CDK inhibitors inhibit apoptosis induced by retinal ischemia-reperfusion, terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) was performed as previously described.25,27 We counted the number of TUNEL-positive nuclei in GCL and INL for a length of 1 mm on either side of the optic nerve head beginning approximately 1 mm from the center of the optic nerve head manually using a light microscope.
For immunohistochemical analysis, tissue sections were rehydrated and subsequently treated for antigen retrieval by boiling samples for 10 min in 10 mM sodium citrate (pH 6.0). Sections were blocked with 10% normal goat serum in PBS, soaked in methanol with 3% H2O2 for 20 min, and then incubated in anti-p16INK4 antibody (1:100; Santa Cruz), anti-p27KIP1 antibody (1:100; Santa Cruz), or anti-CDK4 antibody (1:100; Santa Cruz) overnight at 4°C. Next, sections were treated with histfine simple stain rat MAX-PO (Multi) secondary antibody (Nichirei Bioscience) for 30 min at room temperature. Finally, sections were treated with simple stain DAB solution (Nichirei Bioscience) to observe immunopositive signals. Nuclear counterstaining was performed with methyl green solution.
Drug injection
Roscovitine and a CDK4 inhibitor (2-Bromo-12,13-dihydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione) were purchased from Sigma (St. Louis, MO) and Carbiochem (Darmstadt, Germany), respectively. These drugs were dissolved in dimethyl sulfoxide (DMSO) and diluted by saline. The final concentration of DMSO is 1%. The drug solution was intravenously (i.v.) administered 15 min before 60 min of retinal ischemia. Intravenous injection was performed through a cannula indwelling in the femoral vein.
Statistical analysis
The data represent the means±standard error of the mean. One-way analysis of variance followed by Tukey-Kramer test was used for multiple comparisons. Differences were considered to be statistically significant when the P values were less than 0.05.
Results
Immunohistochemical analysis for cell cycle-related proteins in the rat retina
We determined localization of p16INK4 (Fig. 1B), p27KIP1 (Fig. 1C), and CDK4 (Fig. 1D) in the normal retina. The positive signals for p16INK4, p27KIP1, and CDK4 were seen in all of the retinal layers. No positive staining was seen when nonimmune control IgG was used for the primary antibody (Fig. 1A).

Representative photomicrographs of immunohistochemistry showing distribution of p16INK4
Change in the expression levels of cell cycle-related proteins by ischemia-reperfusion
To determine if ischemia-reperfusion affects the expression levels of p16INK4, p27KIP1, cyclin D1, and CDK4, Western analysis was carried out. Immunoblots showed that the expression levels of p16INK4 and p27KIP1, cyclin inhibitors, were significantly reduced in the ischemic-reperfused rat retinas (24 h after 60 min of ischemia). In contrast, the levels of cyclin D1 and CDK4, which induce cell cycle progression, were clearly increased in the ischemic-reperfused rat retinas (Fig. 2). No positive band was seen when nonimmune control IgG was used for the primary antibody (data not shown).
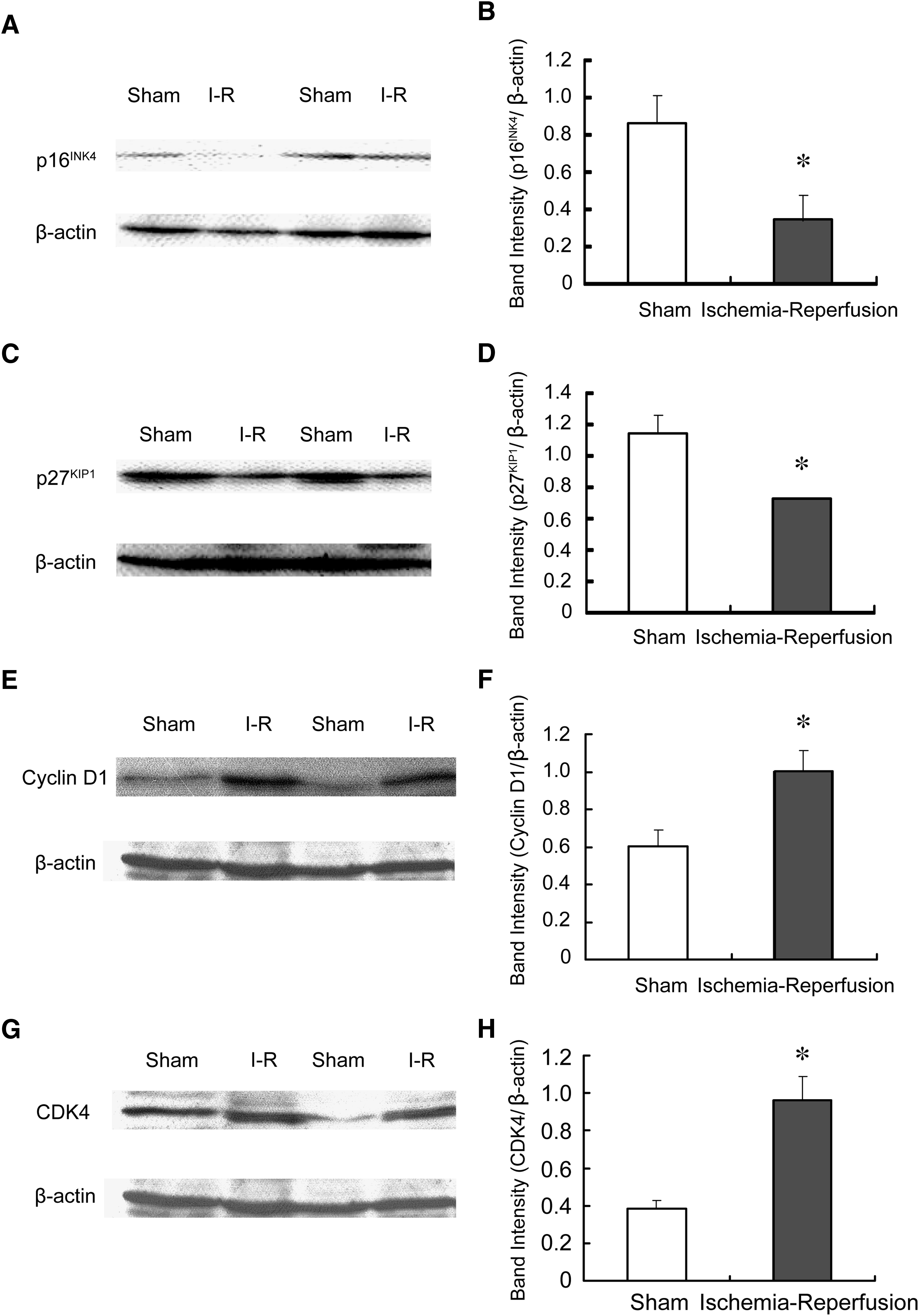
Representative Western blots showing the protein levels of p16INK4
Protective effects of small-molecule CDK inhibitors on ischemic-reperfusion injury in the rat retina
In the ischemic-reperfused retina, cyclin D1 and CDK4 were upregulated and p16INK4 and p27KIP1 were downregulated, suggesting that abnormal cell cycle reentry is involved in the mechanism of cell death induced by ischemia-reperfusion in the rat retina. Therefore, we tested whether small-molecule CDK inhibitors that will block cell cycle progression and/or nonmitotic CDK5 reduced the ischemia-reperfusion injury in the rat retina. Degenerative changes were observed in the GCL, IPL, and INL of the ischemic eye, a characteristic of retinal ischemic atrophy (Fig. 3B), but such changes were not seen in the contralateral nonischemic retina (Fig. 3A). Preischemic treatment with roscovitine (3 mg/kg, i.v.) partially reduced the RGC loss 7 days after 60 min of ischemia (Fig. 3D, G). Pre-ischemic treatment with CDK4 inhibitor (1 mg/kg, i.v.) reduced RGC loss and the decrease of the thickness of IPL 7 days after 60 min of ischemia (Fig. 3F, G).
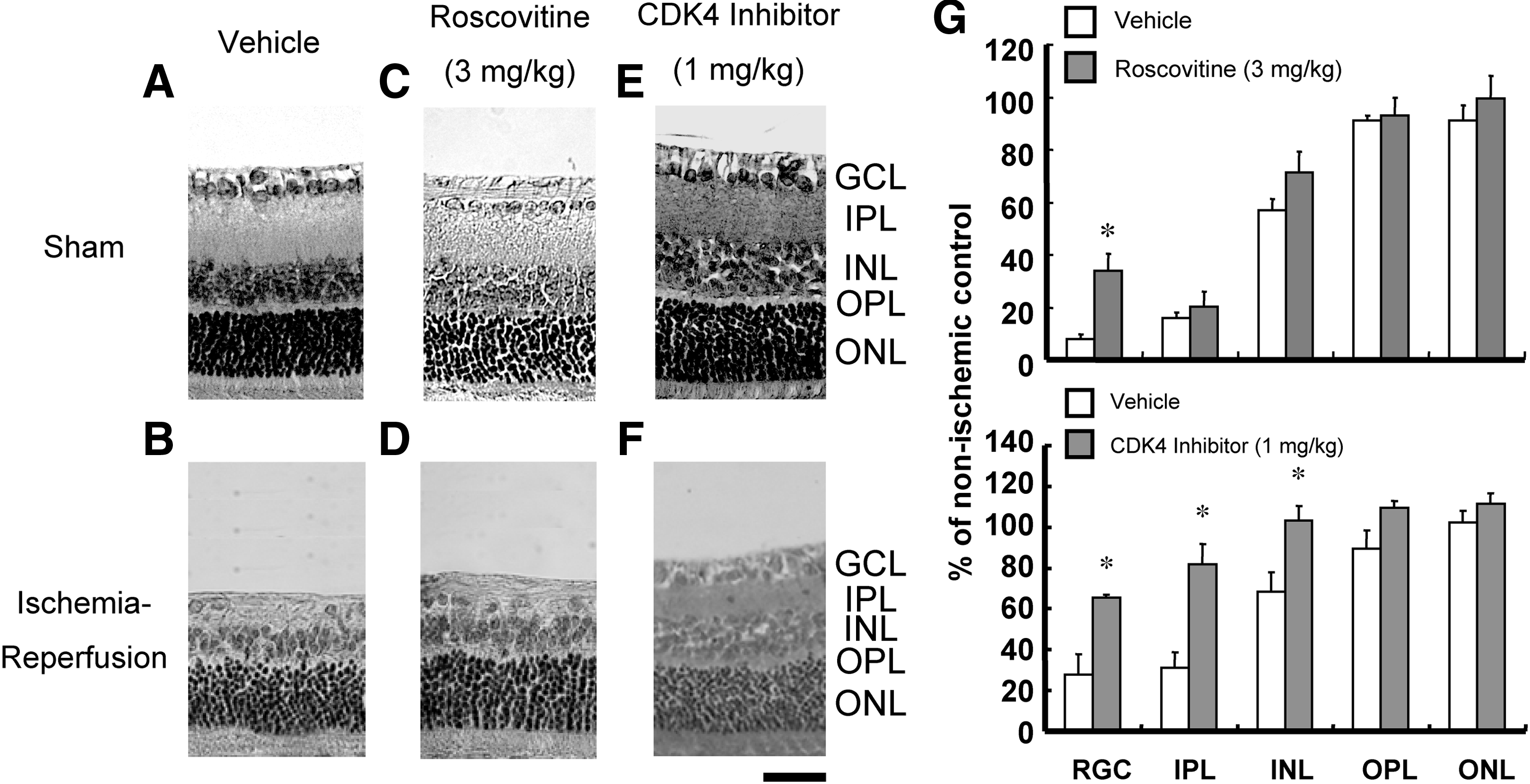
Representative photomicrographs showing the histological appearance of the sham control with the vehicle injection
To examine whether small-molecule CDK inhibitors reduces apoptotic cell death, resulting from retinal ischemia-reperfusion, we conducted TUNEL staining of the retina 6 h after 60 min of ischemia. As shown in Fig. 4, TUNEL-positive cells were observed in the GCL and in the inner side of the INL, but not in the ONL, in the vehicle-treated group. Results of cell count in GCL and INL are shown in Fig. 3G. Preischemic treatment with roscovitine (3 mg/kg, i.v.) and CDK4 inhibitor (1 mg/kg, i.v.) reduced the number of TUNEL-positive cells.
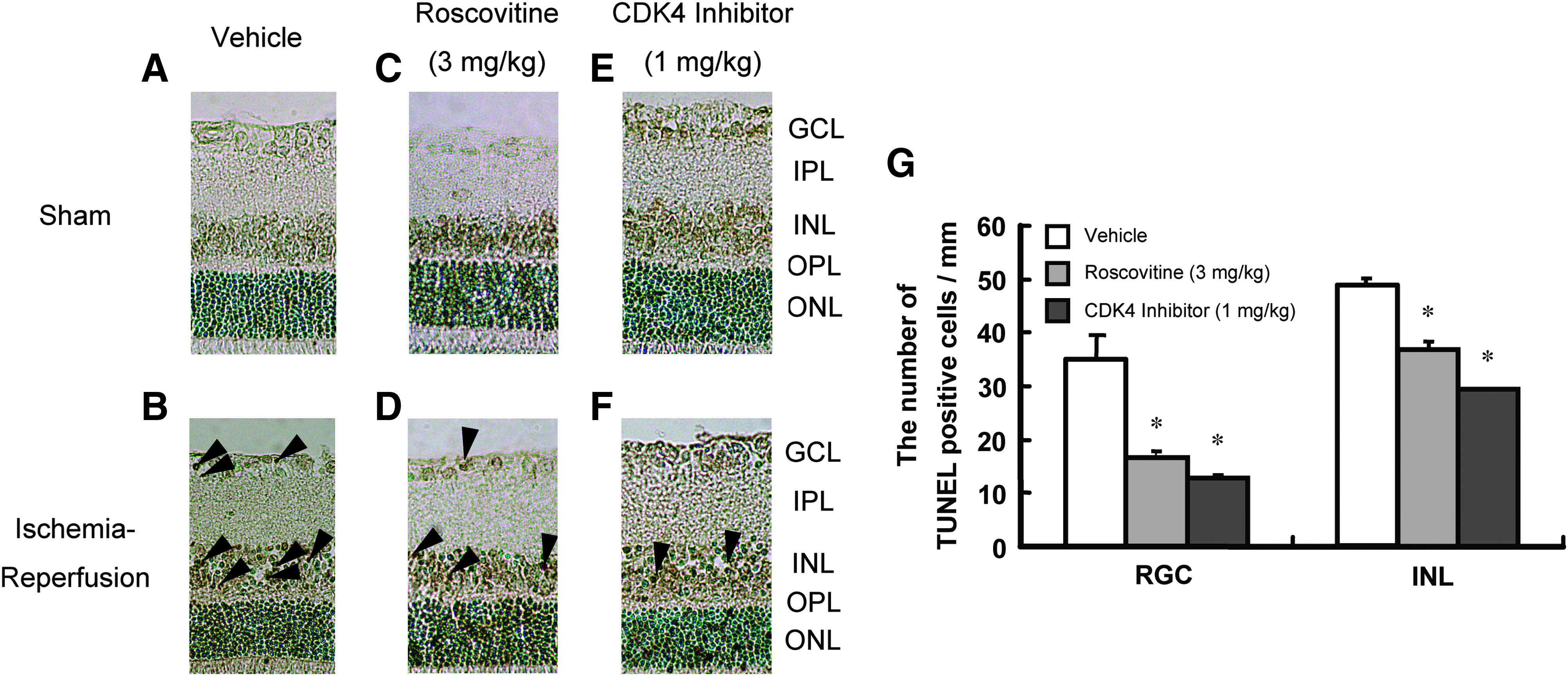
Representative photomicrographs of TUNEL stainings. In the sham retinas
Discussion
Many researchers have thought that the reentry of quiescent neurons into cell cycle may result in mitotic aberration and cell death.6,7,10,12 Cell cycle-related proteins, such as cyclins and CDKs, are reported to be re-expressed and/or to be upregulated in neurons committed to death in response to a variety of insults, including excitotoxins,6,7,28 ischemia-reperfusion,6,7 and β-amyloid peptide. 8 Therefore, activation of cyclins and CDKs can be one of the mechanisms of the induction of neuronal death.
In the present study, we demonstrated that p16INK4, p27KIP1, and CDK4 were constitutively expressed in the normal retina. The data suggest that these proteins expressed in both neurons and other type of the cells, such as Müler glia and microglia. Although we could not clarify the expression pattern of cyclin D1 in the retina, the previous report showed that cyclin D1-positive neurons were seen in the ischemic-reperfused retina, especially in dying cells in INL. 29 It has been shown that p16INK4 positive-horizontal cells were relatively resistant against ischemia-reperfusion injury. 30 We speculate that such strong expression of p16INK4 prevents CDK4/cyclin D1 complex from inducing the reentry to G1 phase in the normal rat retinal neurons. Ischemia-reperfusion significantly increased expression levels of cyclin D1 and CDK4, and decreased those of p16INK4 and p27KIP1 in the rat retina. Such changes in expression of cell cycle-related proteins may release the hook of CDK inhibitors, such as p16INK4 and p27KIP1, and cause cell cycle reentry in the ischemic-reperfused retinal neurons. We cannot exclude involvement of cell proliferation of other cell types, such as microglia, in the results of the present study. Cell cycle proteins were reported to express in proliferating microglia and astrocytes after transient global cerebral ischemia in the rat. 31 Further experiments are needed to clarify the involvement of other cell types in the upregulation of cyclin D1 and CDK4.
To strengthen our hypothesis described above, we tested whether pharmacological suppression of the cyclin-CDK complex could reduce retinal ischemia-reperfusion injury. Roscovitine and CDK4 inhibitor significantly protected against the retinal damage 7 days after ischemia, and the protective effect of CDK4 inhibitor was clearly stronger than that of roscovitine. Because the protective effects of the 2 compounds are clearly different, it is unlikely that reduction of proliferation of other cell types, such as microglia, contributes to the protective effects. In addition, aminoguanidine, which reduces the activation of microglia, did not affect the ischemia-reperfusion injury in the rat retina (data not shown). Therefore, we believe that cell cycle reentry in neurons is one of the mechanisms of ischemia-reperfusion injury in the rat retina. It is known that roscovitine preferentially blocks CDK2 and CDK5 compared with CDK4, 32 whereas there is no information about the inhibitory effect of the CDK4 inhibitor used in the present study on CDK5. Because nonmitotic CDK5 is shown to be associated with neuronal injury induced by cerebral ischemia-reperfusion 21 and axotomy-induced of RGC death, 20 it is possible that inhibition of CDK5 is also involved in the protective effect of these drugs. We think that the difference of the protective effect between the 2 small molecule CDK inhibitors may be caused by substrate specificity.
In the present study, we cannot take the confident proof that the neurons that reenter cell cycle proceed from G0 phase to G1 or M phase. However, we cannot detect bromodeoxyuridine uptake at several time points during reperfusion in the retina (data not shown). Although further detailed experiments are needed, we hypothesize that the neurons that reenter cell cycle do not proceed to M phase through G1 checkpoint.
Various drugs, such as an NMDA antagonist, 33 Ca2+ channel blockers,25,34,35 and nitric oxide synthase blockers, 33 protect the retina in the experimental glaucoma models, including the retinal ischemia-reperfuson model. However, there is no neuroprotective drug to bear practical application for treatment of glaucoma and retinal vessel occlusion. Our findings suggest that inhibition of CDK by small molecules is a new candidate for treatment of retinal diseases known to be associated with excitotoxicity such as retinal vessel occlusion and glaucoma.
Footnotes
Acknowledgment
This work was supported by the Ministry of Education, Science, Sports and Culture, Grant-in-Aid for Young Scientists (B) #17790174 (K.S.) and #21790249 (K.S.), and Kitasato University Research Grant for Young Researchers (K.S.).
Author Disclosure Statement
No competing financial interests exist.