
Abstract
Abstract
Purpose:
This clinical trial evaluated the safety and effectiveness of bepotastine besilate ophthalmic solutions 1.0% and 1.5% compared with placebo for the treatment of ocular itching and conjunctival hyperemia (redness) using the conjunctival allergen challenge (CAC) model of allergic conjunctivitis when dosed 16 h before a CAC test.
Methods:
Subjects with a history of allergic conjunctivitis were assigned to receive placebo or bepotastine besilate ophthalmic solution 1.0% or 1.5% in a single-center, randomized, placebo-controlled clinical trial. Eligible subjects (n=107) aged 10 years and older with a history of allergic conjunctivitis who had a reproducible positive reaction to a CAC were enrolled and dosed with test agent. The primary trial objectives included assessment of ocular itching and conjunctival redness at 16 h after instillation of test agent. Reductions in several CAC-induced secondary symptoms and signs of allergic conjunctivitis were also evaluated for tearing, ciliary and episcleral redness, eyelid swelling, chemosis, and mucous discharge.
Results:
Bepotastine besilate ophthalmic solution 1.5% demonstrated clinical effectiveness and statistical significance in comparison to placebo for the reduction in CAC-induced ocular itching 16 h after drug administration. Bepotastine besilate ophthalmic solution 1.0% also achieved statistical significance in comparison to placebo for reducing ocular itching at all time points 16 h after dosing. Statistically significant reduction (P≤0.05) was additionally seen in this CAC test for the secondary ocular efficacy variable of allergen-induced tearing for bepotastine besilate ophthalmic solution 1.5%. No clinical benefit was seen for reducing the coprimary efficacy variable of conjunctival redness with the CAC model of allergic conjunctivitis.
Conclusions:
Bepotastine besilate ophthalmic solution 1.5% produced predefined clinically meaningful reduction in CAC-induced ocular itching and tearing in a single-site trial and was more effective than bepotastine besilate ophthalmic solution 1.0% and placebo for reducing ocular itching in a CAC test 16 h after dosing.
Introduction
Antagonism of histamine–receptor interactions is frequently used to treat ocular allergies, which leads to improvement in ocular itching and in redness, decreased swelling of the eyelids and conjunctiva, as well as reduction in watery eyes due to allergen exposure. These favorable effects can be provided by antihistamine medications formulated for ophthalmic use. The latest-generation ophthalmic antihistamines are noted for having multiple mechanisms of action 4 that may allow for a longer duration of activity than older antihistamines and fewer side effects.
Bepotastine besilate is one of the newer generation antihistamines, and is a selective H1 receptor antagonist and mast cell stabilizer with demonstrated preclinical and clinical effectiveness.5–7 Bepotastine has been shown to stabilize mast cells,7–10 inhibit eosinophil migration,5,6 block the synthesis and release of newly formed mediators such as leukotrienes7,11 and interleukin-5,12,13 and reduce the inflammatory effects of exogenously applied mediators such as histamine and platelet activating factor. 8 The clinical effectiveness and safety of bepotastine besilate as an oral medication for treating multiple allergic conditions were demonstrated in multiple clinical trials in Japan.14–20 In 2000, an oral formulation of bepotastine besilate was approved in Japan for the treatment of allergic rhinitis, and in 2002 the indication was expanded to the management of urticaria and pruritus associated with skin diseases. 21 Evaluation of the clinical effectiveness and safety of bepotastine besilate as an ophthalmic solution using the well-established conjunctival allergen challenge (CAC) clinical model of allergic conjunctivitis subsequently has been conducted.22,23 As a result of positive outcomes in CAC trials and a favorable safety profile, bepotastine besilate ophthalmic solution 1.5% was recently approved in the United States for treating itching associated with the signs and symptoms of allergic conjunctivitis when dosed twice daily.
The CAC is a standardized and reproducible clinical method of inducing an ocular allergic reaction and evaluating the effects of ophthalmic antiallergy medications in a controlled clinical setting. Although the CAC methodology does not directly address the action of compounds as a mast cell stabilizer in human conjunctiva, the CAC has been validated by use in clinical trials with earlier generation ophthalmic agents that were subsequently approved by the U.S. Food and Drug Administration (FDA).24–28 Positive results have been demonstrated in 2 well-controlled CAC clinical trials (one single site, one multisite) for the primary endpoints of ocular itching and conjunctival redness when assessed 15 min and 8 h after instillation of bepotastine besilate ophthalmic solutions,22,23 and for the relief of nonocular allergic symptoms, including nasal congestion and rhinorrhea. 29 Those clinical trial results led to the approval of Bepreve® (bepotastine besilate ophthalmic solution) 1.5% in September 2009 by the FDA for the treatment of ocular itching associated with the signs and symptoms of allergic conjunctivitis. 30
The single-site CAC clinical trial tested the hypothesis that bepotastine besilate ophthalmic solution 1.0% or 1.5% may reduce the primary efficacy variables of CAC-induced ocular itching and conjunctival redness with sustained clinical tolerability compared with placebo for at least 8 h duration. An additional primary aim addressed in this article was to examine any prolonged beneficial effects seen in the single-site CAC clinical trial 16 h after dosing of bepotastine besilate ophthalmic solutions 1.0% and 1.5% compared with placebo for treating ocular allergic responses. It was postulated that successful demonstration for reduction in CAC-induced ocular allergic responses at 16 h postdosing with bepotastine besilate ophthalmic solution 1.0% or 1.5% might potentially support once daily dosing by allergic conjunctivitis patients.
Methods
Clinical trial design and medications
This was a single-center, double-masked, randomized, placebo-controlled clinical trial. The first 2 clinical trial visits were screening visits to identify subjects with a reproducible and consistent response to an ocular test allergen. Efficacy was then determined by inhibition of the allergic reaction induced by a CAC test 16 h, 8 h, and 15 min after test agent instillation at successive trial visits.
The test agents were placebo (vehicle), bepotastine besilate ophthalmic solution 1.0%, and bepotastine besilate ophthalmic solution 1.5%. Both bepotastine besilate formulations and placebo were identically supplied and labeled and were double-masked as to bottle content from subjects and investigators.
Clinical trial requirements/guidelines
The clinical trial protocol and informed consent form were approved by an independent institutional review board (IntegReview, Austin, TX) before initiation of the clinical trial. The clinical trial was registered on the clinicaltrials.gov Web site (NCT00424398). All trial visits for enrolled subjects were performed in a single outpatient ophthalmic clinic setting (Andover Eye Associates, Andover, MA) between March 1, 2007, and April 4, 2007. Before any clinical trial procedures, a signed informed consent (and assent if the subject was <18 years of age), and a signed Health Insurance Portability and Accountability Act form were obtained from all subjects. The clinical trial was conducted in accordance with International Conference on Harmonisation Good Clinical Practice guidelines, the World Medical Association Declaration of Helsinki (1996 version), and applicable regulatory requirements. This was a prospective, placebo-controlled CAC clinical trial. All ophthalmic examinations and trial-related procedures were performed by clinically trained researchers with experience with CAC methodology.
Criteria for clinical trial participation
Subjects had to meet the following inclusion criteria to be eligible for participation in the clinical trial: (1) be at least 10 years of age and of either sex and any race; (2) have provided written informed consent (and assent if applicable); (3) have negative pregnancy test from all women of childbearing potential, as well as a medically acceptable form of birth control for the designated period; (4) have a positive history of ocular allergies and a positive skin test reaction to cat hair, cat dander, grasses, ragweed, and/or trees within the past 24 months; (5) have best-corrected visual acuity of 0.7 logMAR or better using an Early Treatment Diabetic Retinopathy Study (ETDRS) chart; (6) have positive bilateral CAC reaction within 10 min of allergen instillation at visits 1 and 2; and (7) avoid disallowed medication and contact lens wear for the designated period.
The presence of any of the following conditions prohibited entry into the trial: (1) known contraindications to trial medication(s) or their components; (2) any systemic or ocular condition that could have affected a subject's safety or trial parameters; (3) ocular surgery within 3 months of visit 1 or refractive surgery within the past 6 months; (4) signs of active allergic conjunctivitis at the start of any visit; (5) use of disallowed topical or systemic medications during the designated period; (6) use of an investigational drug or device within 30 days of the trial or concurrently enrolled in another trial; and (7) being a woman who was pregnant, planning a pregnancy, lactating, or not using a medically acceptable form of birth control for the designated period.
Subjects who met these initial entry criteria and did not meet any exclusion criterion at visit 1 then underwent trial screening procedures.
Clinical trial visits
Visit 1 (day −21±3): allergen titration CAC
Eligible subjects underwent a titration CAC with 1 drop of allergen (e.g., Timothy grass or cat dander) instilled bilaterally in the conjunctival cul de sac at the weakest dilution. If the subject failed to react within 10 min, increasingly concentrated doses of the allergen were instilled bilaterally at 10-min intervals until a positive reaction was elicited (score of ≥2 for ocular itching and ≥2 for redness in the conjunctival vessel bed in each eye). Subjects who did not meet this qualification were discontinued from screening. Upon completion of the CAC, an ocular examination was performed on each subject that included distance visual acuity using an ETDRS vision chart, slit lamp biomicroscopy, intraocular pressure, and funduscopy through dilated pupils.
Visit 2 (day −14±3): allergen confirmation CAC
A confirmatory CAC was performed bilaterally with the same allergen and concentration that elicited the adequate allergic response at visit 1. Subjects who reacted positively in both eyes as defined for visit 1 for at least 2 out of 3 time points within this 20 min interval continued in the clinical trial; all other subjects were discontinued from the clinical trial. Ocular itching was graded by subjects at 3, 5, and 7 min post-CAC, and conjunctival redness was graded by the investigator at 7, 15, and 20 min post-CAC.
Visit 3A (day 0): 16 h duration of action dosing
After updating a subject's medical and medication history, the distance visual acuity using ETDRS and slit lamp biomicroscopic examinations were performed. Baseline ocular allergic symptomatology also was assessed. Eligible subjects were enrolled into 1 of 3 treatment groups according to a computer-generated randomization code to receive placebo, bepotastine besilate ophthalmic solution 1.0%, or bepotastine besilate ophthalmic solution 1.5%. No attempt was made to stratify the randomization process. A trained technician instilled 1 drop from the subject's double-masked test agent bottle onto each eye. After 15 min, a safety (visual acuity and biomicroscopy) examination was performed in subjects <18 years of age. All subjects were told to return ∼15.5 h after drug instillation for the visit 3B CAC.
Visit 3B (day 1): 16 h duration of action CAC
Sixteen hours (±30 min) after drug or placebo instillation, subjects at visit 3B received 1 drop of the allergen solution bilaterally. Ocular itching was graded by the subjects at 3, 5, and 7 min postchallenge and conjunctival redness was graded by the investigator at 7, 15, and 20 min postchallenge. Grades for secondary ocular efficacy variables (tearing, ciliary and episcleral redness, eyelid swelling, chemosis, and mucous discharge) were also determined.
Details of visit 4 and visit 5 procedures and effectiveness outcomes, including study exit procedures, have been previously described.22,23,29 In brief, subjects received 1 drop of assigned test agent in each eye at visit 4 (day 14±3), and then a CAC test was performed 8 h after the test agent instillation. At visit 5 (day 28), enrolled female subjects capable of becoming pregnant received a second urine pregnancy test. All subjects then received a third and final drop of assigned test agent in each eye. Fifteen minutes after test agent instillation at visit 5, a CAC test was performed and subjects were exited from the clinical trial. Grading of ocular itching, conjunctival redness, and all secondary efficacy variables was conducted for visit 4 and visit 5 as at visit 3B.
Efficacy and safety assessments
Primary ocular efficacy variables were subject-assessed ocular itching and physician-assessed conjunctival redness. Severity scales for both variables were based on a 5-point (0–4 U) standardized scale, with half-unit increments allowed. 25 Tearing and ocular mucous discharge as ocular secondary efficacy variables were graded as present or absent.
Safety assessments included distance visual acuity using an ETDRS chart and slit lamp biomicroscopy performed at the beginning of each visit. For enrolled subjects under the age of 18, these assessments were also performed 15 min postdrug instillation at visit 3A. The intraocular pressure and funduscopy through dilated pupils examinations were conducted after efficacy assessments at visit 1 and at the last trial visit. Treatment emergent adverse events (AEs; reported, elicited, or observed) were recorded at visit 3A and 3B as well as at later study visits.
Data analysis and statistical methods
Demographic and baseline medical history data were summarized descriptively. For quantitative baseline demographic variables, summaries included the mean, median, and standard deviation, and were analyzed using a 2-sided t-test. Qualitative demographic variables were summarized using counts and percentages, and were analyzed using Fisher's exact test.
The prospectively defined primary analysis population was the per protocol (PP) population. Subjects had to attend visit 3A and 3B or visit 4 and also had to attend visit 5 to be considered for the PP population. An alternative analysis population evaluated for significant changes in efficacy outcomes was the intent-to-treat (ITT) population with last observation carried forward (LOCF) for imputation of missing data. The Wilcoxon rank sum test was used to compare outcomes for the primary efficacy variables of ocular itching and conjunctival redness between each active treatment group and the placebo group in the primary analysis population at each observation time point for visit 3B, the CAC test occurring 16 h after instillation of test agents. The significance levels used to assess the P values for the primary efficacy variables were adjusted for multiple comparisons using conservative Bonferroni corrections to recognize multiple bepotastine besilate concentrations and 2 potential duration of actions, efficacy at 16 h assessed at visit 3B or efficacy at 8 h assessed at visit 4. As a predefined result, treatment differences for either primary efficacy variable therefore were considered statistically significant at α (2-sided)≤0.0125 when observed at a majority of observation time points. Since secondary efficacy variables were considered nonvalidated surrogates for possible clinical activity, P values determined for all secondary efficacy variables other than allergen-induced tearing and ocular mucous discharge were calculated by the Wilcoxon rank sum test and were adjusted by the false discovery rate method. 31 Statistical significance for allergen-induced tearing and ocular mucous discharge was evaluated using generalized linear models, treating each subject as a cluster. Statistical significance for all secondary efficacy variables required α (2-sided) ≤0.05 at all observation time points.
A treatment group response considered clinically meaningful required achievement of clinical significance at a majority of observation time points, as well as statistical significance. Clinical significance at each time point was defined as a ≥1.0-U difference between bepotastine besilate ophthalmic solution (1.0% or 1.5%)–treated eyes and placebo-treated eyes, applicable for all primary and secondary efficacy variables except tearing and mucous discharge. For allergen-induced tearing and mucous discharge, clinical significance was equated to statistical significance.
Additional analyses were performed to examine the clinical and statistical significance for the coprimary endpoints of ocular itching and conjunctival redness for the subset of subjects who experienced grade 3 or greater itching at screening visit 2 in 1 or both eyes, or received a total redness score of 4.5 or greater summed for both eyes. Fisher's exact test was used to compare the proportion of subjects from each treatment group who achieved a mean itching grade of 0 to 0.5-U, etc at all post-CAC time points within the PP study population. This analysis was also performed for the subjects who were more severely affected by a CAC, having an itching grade of 3 or greater in 1 or both eyes at any time point at visit 2.
Assuming the standard deviation of the mean scores for ocular itching and conjunctival redness were each 1.0-U for all 3 treatment groups (based on the results of previous CAC studies with ophthalmic antihistamines), a sample size of 23 subjects per group was anticipated to have >85% power at a post-CAC time point to detect a true mean score difference of 1.0-U between any 2 groups using a 2 sample t-test. Given the nonparametric nature of measures for primary and secondary efficacy endpoints, and with the anticipation of an approximate 10% drop-out rate, 30 subjects per treatment group was used as a target enrollment guide.
Results
Subject sample characteristics
A total of 179 subjects were screened and 107 were enrolled and randomly assigned a masked test agent: 36 subjects to each of placebo and bepotastine besilate ophthalmic solution 1.0% and 35 subjects to bepotastine besilate ophthalmic solution 1.5%. Of those enrolled, 102 completed the clinical trial including attendance at visit 3A and 3B, of whom 3 subjects committed protocol violations that removed them from consideration for the PP population. This left 34 subjects in the placebo group, 35 subjects in the bepotastine besilate ophthalmic solution 1.0% group, and 30 subjects in the bepotastine besilate ophthalmic solution 1.5% group comprising the PP population with data for the 16 h duration of action (i.e., visit 3A and 3B). The subject demographics among all subjects were well balanced for bepotastine besilate ophthalmic solution 1.0% and 1.5% compared with placebo for age, gender, race, and iris color (Table 1).
P value for age as a continuous variable was calculated by 2 sample t-test. All other P values were calculated using Fisher's exact test.
SD, standard deviation.
Efficacy variable outcomes
Ocular itching
For reduction in CAC-induced ocular itching, both bepotastine besilate ophthalmic solutions 1.0% and 1.5% achieved statistical significance in comparison to placebo at all time points 16 h after dosing in both the PP population (P<0.001) and the ITT population with LOCF (P≤0.001) with LOCF (Table 2). Bepotastine besilate ophthalmic solution 1.5%, but not bepotastine besilate ophthalmic solution 1.0%, also achieved clinical significance in comparison to placebo in the PP population for the reduction of ocular itching at all time points 16 h after dosing.
Difference in mean itching scores in the PP and the ITT with LOCF populations.
P≤0.001.
CAC, conjunctival allergen challenge; ITT, intent-to-treat; LOCF, last observation carried forward; PP, per protocol.
An analysis was performed to determine the proportion of subjects in each group that evidenced a 2.0-U, etc reduction in itching scores compared with their screening visit scores within the PP population, as well as for the subgroup of subjects in the PP population with the most severe itching (defined as an ocular itch score of 3 or more in each eye at a screening visit 2 time point). Within the PP population, 40.0% of subjects in the bepotastine besilate ophthalmic solution 1.5% group experienced a 2.0-U reduction for at least 1 post-CAC time point at visit 3B compared with 34.3% of those in the bepotastine besilate ophthalmic solution 1.0% group and 5.9% in the placebo group, demonstrating a dose-dependent reduction in itch. Of those in the severe itching subpopulation, a 2.0-U reduction in eye itching scores for at least 1 post-CAC time point was measured in 8.7% of the placebo group compared with 37.5% and 43.5% of the bepotastine besilate ophthalmic solution 1.0% and 1.5% group subjects, respectively (Fig. 1). In both the PP population and severe itching subpopulation, the greater number of subjects that experienced 2.0-U reductions in itching scores with either bepotastine besilate ophthalmic solution (1.0% or 1.5%) treatment compared with placebo was statistically significant (P=0.001 and P=0.008, respectively).

Percent of subjects with 2-U improvement in ocular itching scores (placebo–active) at 1 or more observation time points relative to visit 2 screening values. Changes in ocular itching scores were pair-matched for each subject by eye and observation time point at 16 h after dosing with masked medication. Results for left and right eyes were averaged. PP, per protocol population.
Given that subject enrollment required an ocular itching score of 2.0-U or greater at visit 1 and visit 2, a 2.0-U reduction in itching would be expected to produce near complete elimination of ocular itching. Figure 2 illustrates the proportion of subjects that achieved complete or near complete elimination of ocular itch (score of 0 or 0.5-U) 16 h after dosing at a post-CAC time point. A greater number of subjects receiving either bepotastine besilate ophthalmic solution achieved complete or nearly complete elimination of itch following a CAC compared with placebo in both the PP population (P<0.001) and the baseline severe itching subpopulation (P=0.005). For both study populations, the degree of reduction or elimination of ocular itching was greater in the bepotastine besilate ophthalmic solution 1.5% group relative to results for the bepotastine besilate ophthalmic solution 1.0% group, although the differences were not statistically significant (P>0.05).
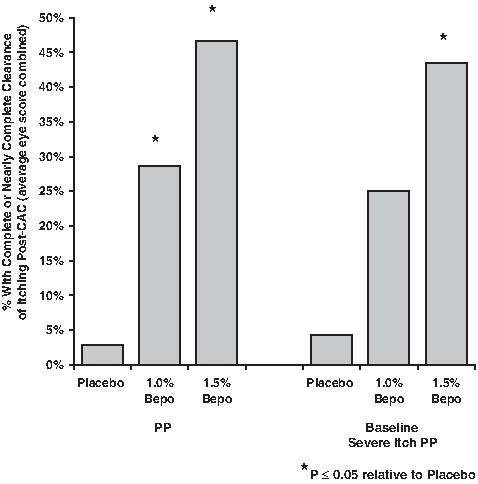
Percent of subjects with complete or nearly complete clearance of itching post-CAC at 1 or more observation time points 16 h after dosing. Results for left and right eyes were averaged. PP, per protocol population.
Conjunctival redness
Bepotastine besilate ophthalmic solution 1.0% was statistically superior (P≤0.012) to placebo for reducing mean conjunctival redness only at the 7 min time point 16 h after dosing in both the PP population and the ITT population with LOCF. However, there were no clinically significant (i.e., ≥1.0-U) differences in conjunctival redness between bepotastine besilate ophthalmic solutions (1.0% or 1.5%) and placebo at any time point 16 h after dosing.
Tearing (lacrimation)
Shown in Table 3 is the proportion of subjects' eyes with allergen-induced tearing absent at all post-CAC time points exclusively for those subjects that had tearing eyes observed during screening visit 2. A dose relationship was found for the relative reduction in allergen-induced tearing measured between screening and 16 h postdosing. The placebo group had a 27.5% reduction in eyes with tearing present at baseline, compared with a 51.2% reduction in the bepotastine besilate ophthalmic solution 1.0% group (P≤0.05 relative to placebo) and an 85.7% reduction in the bepotastine besilate ophthalmic solution 1.5% group (P<0.0001 relative to placebo). There was also a significantly greater reduction of tearing in the bepotastine besilate ophthalmic solution 1.5% group as compared with the bepotastine besilate ophthalmic solution 1.0% group (P=0.0046).
P≤0.05 relative to placebo.
P≤0.0001 relative to placebo.
P≤0.005 relative to bepotastine besilate ophthalmic solution 1.0%. P values calculated using Fisher's exact test.
In general, regardless of whether or not subjects experienced allergen-induced tearing at visit 2, there was less tearing in both bepotastine besilate ophthalmic solution groups as compared with placebo at all 3 post-CAC time points at visit 3B, in both the PP population and the ITT population with LOCF. In the PP population in particular, there were between 3.1% and 9.4% tearing eyes in the bepotastine besilate ophthalmic solution 1.5% group as compared with between 36.8% and 47.1% tearing eyes in the placebo group, and the differences were statistically significant at each time point (P<0.001). Tearing in the bepotastine besilate ophthalmic solution 1.0% group ranged between 20.0% and 24.3%, and also was significantly less than the placebo group 7 and 15 min post-CAC (P≤0.030).
Safety
The safety population (n=107) was defined as subjects who received at least 1 dose of test agent. A total of 5 ocular AEs were reported, 2 originating from the bepotastine besilate ophthalmic solution 1.5% group (eye irritation and conjunctival cyst) and 3 from the placebo group (eye irritation and foreign body sensation). Eighteen subjects reported 29 nonocular AEs (Table 4). Fourteen of these nonocular AEs were classified as related to treatment, reported by 9 subjects. Unpleasant or bitter drug taste was reported upon eyedrop instillation by 6 subjects in the bepotastine besilate ophthalmic solution 1.0% group and by 3 subjects in the bepotastine besilate ophthalmic solution 1.5% group. All 9 subjects reported this event as mild or moderate.
AE, adverse event.
Discussion
Bepotastine besilate ophthalmic solutions 1.0% and 1.5% were both previously reported to be effective for the reduction of ocular itching in this double-masked, single-site trial for at least 8 h using the CAC model of allergic conjunctivitis.22,23 The data analyses presented here for bepotastine besilate ophthalmic solution 1.5% also show clinical success (≥1.0-U improvement relative to placebo; P<0.001) in reducing ocular itching when challenged 16 h after drug delivery, suggesting the possibility of a once daily dosing regimen. It appears that the higher bepotastine besilate concentration provided the potency required to achieve this clinically significant effect for up to 16 h. Subgroup analyses for the present clinical trial revealed a dose-dependent improvement in ocular itching that was statistically significant among subjects with more severe ocular itching scores at screening as well as for all subjects when treated with either concentration of bepotastine besilate ophthalmic solution. This finding has important implications since increased severity of allergic conjunctivitis symptoms can lead to a more refractory therapeutic response. Bepotastine besilate ophthalmic solutions therefore appear to offer an efficacious and rapid therapy for the severely allergic patient, which may allow such severe patients to avoid or reduce steroid use and thereby minimize the detrimental effects associated with those drugs. 32 Based on the analysis of AEs from this trial, both concentrations of bepotastine besilate ophthalmic solution also were safe and well tolerated. Ocular AEs in this clinical trial were minimal and did not include any instances of dry eye.
Results of this clinical trial are comparable to previous clinical trials of bepotastine besilate as a systemic medication in treating itching associated with other allergic conditions.13,16,33–37 Statistical significance for reduced itching was seen in the multicenter CAC trial for both bepotastine besilate ophthalmic solutions (1.0% and 1.5%) compared with placebo for the 16-h CAC test (P<0.0055 at all time points), but a 1.0-U improvement in ocular itching compared with placebo was not demonstrable for either bepotastine besilate formulation and clinical benefit was not seen for reducing conjunctival redness.
Lacrimation is a major problem in patients with allergic rhinoconjunctivitis. A cross-sectional survey of patients (n=447) with allergic rhinitis and their physicians showed that at the time of their consultation, 44.0% of patients were suffering from both nasal and ocular symptoms. 38 Furthermore, 13.6% of patients reported itchy, red eyes, or watery eyes as their most troublesome symptom. 38 An ocular allergy treatment for tearing or watery eyes that did not induce dry eye side effects would address the needs of such patients. The data presented here therefore demonstrate that both bepotastine besilate ophthalmic solutions (1.0% and 1.5%) have possible clinical value by virtue of showing significant activity for reducing allergen-induced excessive tearing or watery eyes in the CAC model.
Sisler et al. 39 reported in 1982 that the lacrimal drainage system included valves assuring the progression of tears in 1 direction; secretions pumped from the lacrimal gland are distributed over the ocular conjunctiva and drain from the ocular surface through the punctum; the punctum carries secretions to the common canaliculus, moving then to the lacrimal sac and nasolacrimal duct, where secretions drain into the nose. This one-way drainage hypothesis is supported by a conjunctival and nasal allergen challenge study suggesting that lacrimal secretions gained access to the nose through the nasolacrimal duct but nasal secretions could not reach the eye. 40 Therefore, it is likely that CAC-induced inflammation causing nasal congestion might block the drainage of tears through the nasolacrimal duct and lead to excessive watery eyes. It is notable that in the present study, the dose-dependent reduction in watery eyes observed 16 h after treatment in subjects receiving bepotastine ophthalmic solution 1.0% and 1.5% correlates with previously published results demonstrating statistically significant reductions (P≤0.01) in nasal congestion and rhinorrhea with bepotastine besilate ophthalmic solution 1.5% 16 h after subject dosing. 29 Further studies appear warranted to better understand the relationship between CAC-induced tear production and the actions of H1 receptor antagonists such as bepotastine besilate ophthalmic solutions.
The CAC model has been accepted by the FDA as a standardized and reproducible means of evaluating the efficacy and duration of action of ophthalmic antiallergic agents.41,42 At a CAC visit, the test agent is instilled into the conjunctival cul de sac of subjects with a history of allergic conjunctivitis. At a predetermined time later, an allergen is instilled to induce an ocular allergic response intended to be similar across all subjects. Changes in the signs and symptoms resulting from the allergen challenge are graded according to standardized severity scales at predetermined time points, allowing for relatively precise comparisons of the effects of ocular allergy drugs among subjects and the reproducibility of effects for any 1 subject. The scales and grading procedures also afford the sensitivity needed for drug onset of action and duration of action clinical studies, in comparison to either placebo or to an active comparator. However, the CAC model does have study limitations. To avoid interference by seasonal allergens, CAC clinical trials are often conducted outside the allergy season. Exposure to high concentrations of different allergens in the CAC model and study designs that emphasize prevention rather than treatment of allergic symptoms may be seen as limitations of the model. However, the more numerous drawbacks inherent in a seasonal allergic conjunctivitis trial, such as day-to-day variability of allergen exposure and differences in intersubject symptoms, are resolved by the use of this model.
In conclusion, this CAC clinical trial demonstrates the potential of once daily dosing with bepotastine besilate ophthalmic solutions. Since patient treatment, adherence, and quality of life are important issues in allergic diseases, the benefits of once daily dosing are considerable. Studies in other therapeutic areas indicate that a key factor in treatment adherence is the prescribed dosing schedule for a drug.43–45 Suboptimal adherence to treatment regimens results in reduced clinical benefits and diminished quality of life for many patients, especially those with persistent allergies, contact lens wearers, and children. Interestingly, patient adherence has been shown to double when drug regimens decrease from twice a day to once a day. 46 Bepotastine besilate ophthalmic solutions 1.0% and 1.5% have been shown in this CAC trial to be safe and effective topical therapies that provide potent relief of ocular itching in the CAC model of allergic conjunctivitis for at least 16 h. Further clinical research to explore alternative dosing regimens with bepotastine besilate ophthalmic solution 1.5%, the approved ophthalmic antihistamine, in patients having allergic conjunctivitis therefore may be warranted.
Footnotes
Acknowledgments
The authors would like to thank Mauricio Muñoz and Randi L. Rohlman of ISTA Pharmaceuticals, Inc., for assistance in editing the article.
Author Disclosure Statement
This clinical trial was funded by a grant from ISTA Pharmaceuticals®, Inc. The authors have made the following financial disclosures: J.I.W., J.A.G., and T.R.M. are employees of ISTA Pharmaceuticals®, Inc.; K.S.K. is an employee of Statistics and Data Corporation; no competing financial interests exist. G.L.T. is an employee of Andover Eye Associates, and P.J.G and M.B.A. are employees of Ora, Inc.; they have no competing financial interests to disclose.