
Abstract
Abstract
Purpose:
Latanoprost is used for the treatment of an increased intraocular pressure (IOP) to prevent the progression of glaucoma. Since the lack of compliance with topical ocular dosing may compromise efficacy, alternate methods of delivery are being sought. A 9-month study was conducted to assess the safety and tolerability of latanoprost-containing subconjunctivally implanted devices.
Methods:
Dutch-belted rabbits were implanted subconjunctivally with up to 5 placebo or drug-loaded devices containing from 50 to 180 μg of latanoprost per device. Study assessment consisted of irritation scoring, clinical signs, ophthalmic exams, IOP, electroretinography (ERG), ocular histology of cohorts at 3 and 9 months postimplantation, and systemic exposure to latanoprost acid.
Results:
The implants were well tolerated, with minimal-to-mild clinical and microscopic ocular findings attributable to either the placebo or drug-loaded devices. Mild conjunctival congestion persisted through week 13 of the study and tended to correlate with the number of devices and presence of drug. Ophthalmic examinations revealed no effects beyond conjunctival surface hyperemia. No effects on the IOP, corneal thickness, or ERG parameters were observed. The lack of changes in the IOP was expected due to the known lack of the IOP-lowering effects in rabbits from latanoprost. Microscopically, implants at the 3-month necropsy were associated with subconjunctival tissue cavities (containing the implants), fibrous encapsulation, and an infiltrate of lymphocytes and macrophages, sometimes as multinucleate cells, into the subconjunctival implant cavity. The drug-containing implants were often associated with inflammatory cell infiltrates, including heterophils (neutrophils), within the implant subconjunctival cavities and adjacent to the implant sites. At the 9-month necropsy, heterophils were no longer common among the inflammatory cell infiltrates; macrophages and lymphocytes persisted; most of the biodegradable implants were fragmented and disintegrating; and fibrovascular proliferation was present within implant luminal remnants. None of the findings were considered adverse. Systemic exposures were above the limit of quantification (0.1 ng/mL) for up to 96 h in the higher-dose groups, consistent with the initial burst phase of compound release.
Conclusion:
Overall, the study supports the safety of the latanoprost-containing subconjunctival device as a means of extended delivery of the antiglaucoma medication. Latanoprost-containing subconjunctival implants were well tolerated by Dutch-belted rabbits for up to 9 months. Such devices may improve patient compliance and serve as a means of extended delivery of antiglaucoma medications.
Introduction
Prostaglandin F2α analogs (PGA), including latanoprost, which decreases the IOP by increasing the outflow of the aqueous humor, 2 are becoming the preferred choice for first-line treatment of increased IOP, demonstrating an exceptional efficacy and safety profile. 3 As monotherapy, these drugs combine the unsurpassed IOP-lowering efficacy in the most common forms of glaucoma with a unique duration of action allowing once-a-day dosing and a lack of systemic side effects. In addition, PGAs can be combined with most other mode-of-action drugs for additive IOP-lowering effects. The most common ocular side effects of PGA treatment are cosmetic in nature and include conjunctival hyperemia, elongation and darkening of eyelashes, induced iris darkening, and periocular skin pigmentation. 4
Despite the tolerability and potential efficacy of topical ocular drop administration, the lack of adherence to treatment regimens can be an issue in maintaining the control of IOP. Studies have demonstrated that as much as 45% of patients report some degree of nonadherence. 5 Some patients may have increased difficulty adhering to the administration of eye drops, including physical conditions that affect their ability to accurately apply drops to the eye. One possible solution to the lack of adherence is through the use of an extended-release device that would deliver an IOP-lowering drug near the site of action. Due to their potency and low daily dose relative to therapies utilizing other modes of action, PGAs are particularly qualified for the use in extended-release device therapy.
In this report, we studied the safety and tolerability of a subconjunctival implanted device designed for the controlled release of latanoprost. Multiple devices were inserted into the conjunctiva of Dutch-belted rabbits, followed by observation periods of 3 and 9 months, during which time ophthalmic examinations were conducted at designated intervals before necropsy and histological evaluation. Other than the transient initial localized trauma associated with the device insertion procedure, the latanoprost-containing implants were well tolerated during the duration of the study.
Methods
Implants
Devices containing latanoprost, the isopropyl ester prodrug of the FP2α prostaglandin receptor agonist latanoprost acid, were used in these studies. The prodrug is the ingredient found in commercial formulations, has greater bioavailability than the acid, and is rapidly converted to the active acid form by tissue and plasma esterases.
Two types of implantable rod-shaped devices were used in the study (X50A and X190A; pSivida, Watertown, MA), with a length of 3.5 mm and outer diameters of 0.18 and 0.37 mm, respectively, and differing by the inner core (luminal) diameter, shell composition, drug-loading capacity, and biodegradable characteristics. X50A devices had an inner core diameter of 0.15 mm and contained ∼56 μg of latanoprost with an in vitro release rate of 0.24 μg/day and had a shell made of a nonbiodegradable polyimide material. The X190A devices had an inner core diameter of 0.28 mm and contained ∼175 μg of latanoprost with an in vitro release rate of 1.4 μg/day and a shell made of biodegradable poly-lactic-co-glycolic acid (95:5 ratio). The purpose of testing 2 different-sized implants with differing compositions was to maximize the efficiency of the study by enabling the testing of multiple variables (release rate and tolerability of the composition), which was possible since the rate of release is dependent on the inner shell diameter and independent of the composition. Latanoprost was formulated in fumed silica (Cap-o-sil; Cabot Corporation, Alpharetta, GA) as a thickening agent before loading into the devices. Both ends of the devices (release ports) were coated with release-rate-controlling polymer membranes. The placebo devices were identical to the test articles in size, excipients, and polymer materials, except there was no active drug substance in the drug core. A 1-mL modified syringe and needle were used to implant the devices (Medidur; pSivida).
Before implantation, representative samples of each device were tested for their release rate in vitro. Briefly, each implant was placed in a microcentrifuge tube, and 1 mL PBS was added to the tube. The tubes were kept in water bath at 37°C. The samples were taken, and the entire medium was replaced with a fresh buffer every 24 h. Since the release medium for the in vitro release test was replaced every 24 h and the daily release amount of latanoprost is at least 10 times lower than the latanoprost solubility in PBS at 37°C, the sink condition was maintained through the entire test period. The amount of latanoprost released in each 24-h period during the 90-day study was quantitatively measured using a computerized high-performance liquid chromatography (HPLC system equipped with an UV detector capable of monitoring absorbance at 200 nm, an auto-injector, and a column oven). The analytical column was a Nucleosil C18 column (150×3.2 mm, 5μ), and the column was maintained at 60°C. The mobile phase was a mixture of acetonitrile (50%) and 25 mM phosphate buffer (50%). The concentrations of latanoprost were determined by peak area against the latanoprost standard calibration curve.
Animals
The rabbit study was conducted in accordance with the U.S. Food and Drug Administration Good Laboratory Practice Regulations, Title 21 of the US. Code of Federal Regulations Part 58, and any applicable amendments.
Five-month-old Dutch-belted rabbits (Myrtle Rabbitry, Inc., Thompson Station, TN) were randomly assigned to 4 groups (6/sex/group; Table 1). Injections were performed under general anesthesia. Group 1 (control) received no treatment other than the aseptic preparation of the ocular surface for injection as in the other groups. The right eyes of Group 1 received the aseptic preparation of the ocular surface only. The left eyes of animals in Group 1 were aseptically prepared and underwent a sham injection consisting of inserting the needle into the subconjunctival space and withdrawing it. Group 2 received 2 X50A devices in the right eye and 2 X50A placebo devices in the left. Group 3 received 2 X190A devices in the right eye and 2 X190A placebo devices in the left. Group 4 received 5 X190A devices in the right eye and 5 X190A placebo devices in the left. The devices were implanted using the injector described above. The needle was inserted into and under the conjunctiva ∼2–3 mm from the limbus near the superior temporal quadrant. The syringe was directed parallel to the limbus, and the devices were injected under the conjunctiva. Rabbits were necropsied (3/sex/group) on day 93 or day 275. The rabbit study was conducted in adherence to the Association for Research in Vision and Ophthalmology (ARVO) statement for the Use of Animals in Ophthalmic and Vision Research.
Cohort 1 (3/sex/group) animals were euthanized and necropsied on day 93 of the dosing phase (interim euthanasia). Cohort 2 (3/sex/group) animals were euthanized and necropsied on day 275 of the dosing phase (terminal euthanasia).
For Group 1, the right eye remained untreated, and the left eye underwent a sham injection (needle inserted and withdrawn), but no devices were implanted. The sham injection mimicked the procedure done for Group 4 animals. For Groups 2 through 4, the right eye of each animal was dosed with implant devices containing test article, and the left eye received the same number of empty implant devices.
Test Article A=X50A implant device with latanoprost, Test Article B=X190A implant device with latanoprost, Placebo A=empty X50A implant device, and Placebo B=empty X190A implant device.
Ocular endpoints
Ophthalmic examinations using a slit-lamp biomicroscope and indirect ophthalmoscope were conducted during the predose phase and on days 1 (predose and immediately after dosing), 3, 8, 15, and 29, and during weeks 9, 13, 20, 26, and 39. The eyes were examined by slit-lamp, and irritation was scored by a Diplomate of the American College of Veterinary Ophthalmologists using the McDonald–Shadduck scoring system, 6 which had been modified to also note whether inflammatory cells were present in the anterior chamber or vitreous. Representative ocular photographs were taken from selected animals with a hand-held digital camera at the 3-, 6-, and 9-month time points. The IOP was measured using a Tono-vet-rebound tonometer (Acrivet, Henningsdorf, Germany) once during the predose phase, once on day 1, and during subsequent ophthalmic exams. Pachymetry was performed using an ultrasound pachymeter (AccuPach VIR; Accutome, Inc., Malvern, PA) once during the predose phase, on day 1, and during ophthalmic exams thereafter. Electroretinography (ERG) was performed once during the predose phase and in weeks 13 and 38. Scopic tests were done using the following stimuli: dim rod-isolating flash (Scotopic-24 dB white single flash), intermediate luminance (Scotopic-16 dB), mixed rod–cone bright flash (Scotopic 0 dB), and oscillatory potentials (digitally filtered from the Scotopic 0 dB white single flash).
Histology
Upon necropsy on days 93 or 275, the eyes (including bulbar conjunctiva) and optic nerves were preserved by immersion in 6% glutaraldehyde, followed by immersion in 10% neutral-buffered formalin after trimming, but before processing. Extraocular muscles (rectus dorsalis, rectus lateralis, and superior oblique), optic nerves (longitudinal and cross-sectional samples), eyelids (upper and lower, including palpebral conjunctiva), nictitating membranes, and the Harderian and lacrimal glands were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. These tissues were microscopically analyzed by a Diplomate of the American College of Veterinary Pathologists. Devices were left in situ during histological processing.
Pharmacokinetics
Serial blood samples were collected for plasma from individual animals at the following time points: 1, 6, 24, 48, 96, and 312 h, as well as 14, 25, and 41 weeks after dosing on study day 1. These samples were analyzed for latanoprost acid. From the control group, only the zero-hour postdose samples were analyzed. Rabbit plasma was analyzed for latanoprost acid using a validated LC/MS/MS method, and the electronic bioanalytical data were stored in Watson LIMS (v7.2; Thermo, Inc., Philadelphia, PA). All calculations were performed with a validated copy of Phoenix™ WinNonlin® 6.1 (Mountain View, CA), and the mean data were used in all pharmacokinetic (PK) analyses. The area under the plasma latanoprost acid concentration–time curve [AUC(0–96)] was estimated using the linear trapezoidal rule. In the calculations of AUC(0–96), plasma concentrations of latanoprost acid at time 0 were set to 0 ng/mL on day 1. For subsequent dosing days, the plasma concentrations of latanoprost acid at time 0 were set to the corresponding 24-h concentration value for the previous day. Concentrations of 0 ng/mL were used for PK calculations for all results that were below the limit of quantification (BLQ; 0.1 ng/mL).
Results
In vitro
The in vitro latanoprost release rates are shown in Fig. 1. The cumulative latanoprost release for the low-dose (Fig. 1A) and high-dose (Fig. 1B) devices appears linear after the first week until the last time point measured at 90 days. Additionally, ≥50% of the total contents of the devices was released within the 90-day time period. The daily release profiles show an initial burst during the first few days, followed by a relatively constant release rate of 0.24 μg/day for the low-dose device (Fig. 1C) and 1.4 μg/day for the high-dose device (Fig. 1D). These rates would hypothetically translate to 0.48, 2.8, and 7 μg/day in the low-, mid-, and high-dose groups, respectively, in the current rabbit study. Additionally, since the approved topical dose of latanoprost is 1.5 μg/eye/day, a single X190A high-dose device theoretically delivers the appropriate daily dose.

In vitro release profiles for latanoprost-releasing devices. Absolute (left-axis) and relative (right-axis) cumulative release of latanoprost from X50A
Ophthalmic exams
Digital photographs of Dutch-belted rabbit eyes with implanted devices are shown in Fig. 2. Three months after implantation, 5 (Fig. 2A) or 2 (Fig. 2B) devices are visible at the site of implantation.
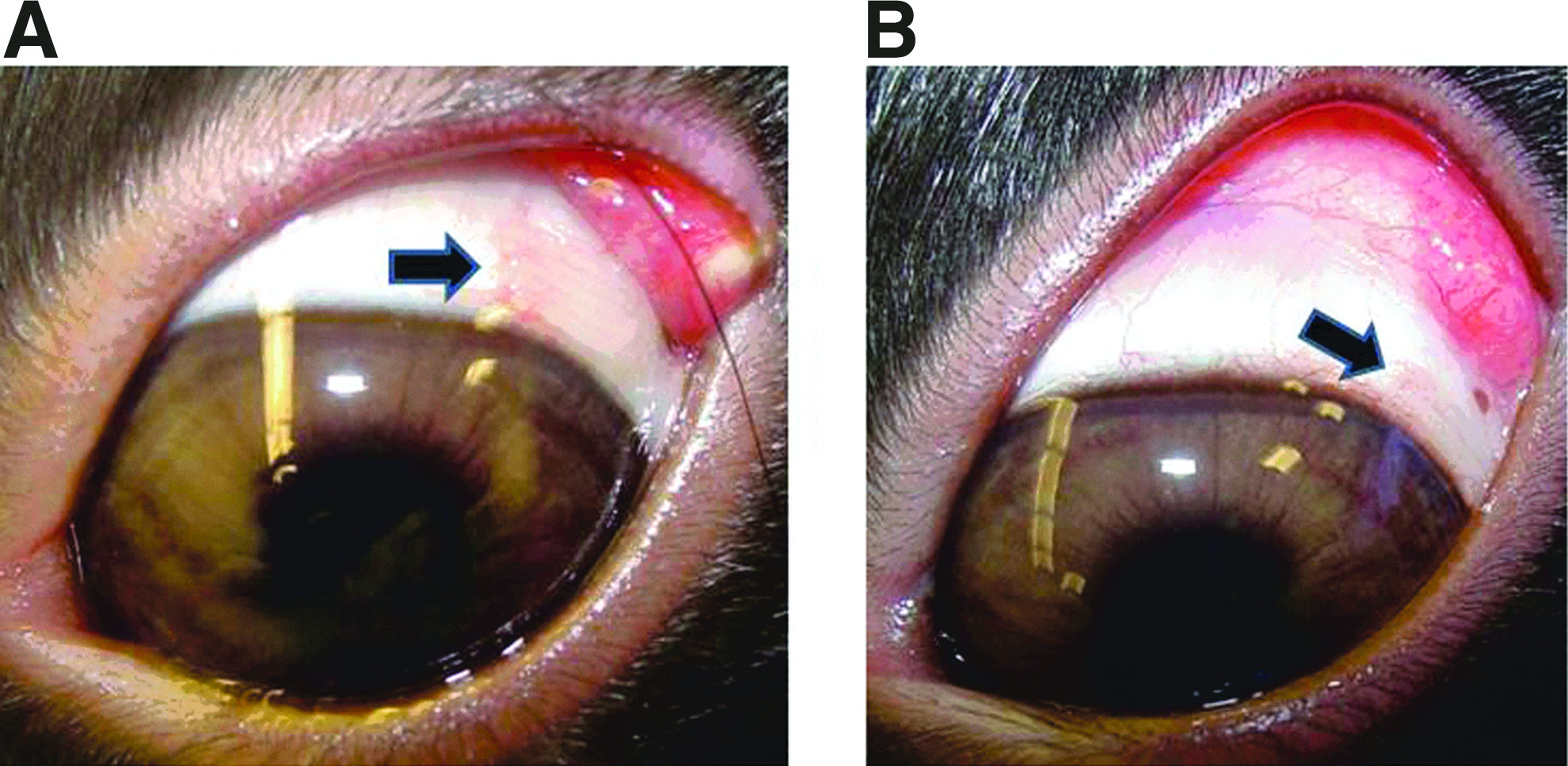
Representative ocular photographs 3 months after implantation of 5
The findings in the ophthalmic examinations were limited to irritation scoring using the McDonald–Shadduck grading scheme. In general, subconjunctival administration of 2 X50A implants and 2 or 5 X190A implants, with or without latanoprost, was well tolerated for 39 weeks in this study. Peak ocular responses occurred on day 3 (Table 2A) of the dosing phase in all groups, including Group 1 controls aseptically prepared with (right eyes) or without sham injections (left eyes), and typically consisted of mild (1+) to moderate (2+) conjunctival congestion (also referred to as conjunctival hyperemia; diffuse unless otherwise noted) and mild conjunctival chemosis. These findings resolved before day 8 (Table 2B) of the dosing phase in all Group 1 control eyes and were principally attributed to the sterile preparation of the ocular surface, grasping of the conjunctiva for injection, and possible minor ocular surface drying or trauma upon recovery from general anesthesia. Based on comparisons between the eyes that had only been aseptically prepared (right eye, Group 1) and the eyes that had been aseptically prepared and also received a sham subconjunctival injection (left eye, Group 1), the injection itself contributed negligibly to this response. Usually, mild conjunctival congestion was variably present after day 3 of the dosing phase in the eyes given any implant (with or without latanoprost), and as the number or size of implants increased, this generally tended to be more frequent or slightly higher in score through week 13 (Table 2C) of the dosing phase, but not thereafter. Additionally, the eyes given any implant containing latanoprost tended to exhibit mild (1+) conjunctival congestion localized to the injection site at a greater frequency than that seen in eyes given a comparable size or number of placebo implants through weeks 9 to 13, but typically not thereafter. In the aggregate, these findings suggest that the mild conjunctival congestion seen between day 3 and week 13 in eyes given implants was due in part to the size and number of implants as well as the dose of latanoprost delivered. After week 13, this relationship was no longer clearly discernable. Over the weeks of 20, 26, and 39, the incidence of minimal conjunctival congestion, which was observed in the majority of mid- and high-dose animals at week 20, decreased in incidence over time and was similar between right and left eyes.
There were no other treatment-related findings in other ophthalmic measurements, including corneal thickness, ERGs, slit-lamp examinations, or IOP (data not shown).
Histology
Microscopic observations: 3-month (interim) necropsy
Postimplantation left and right eyes from control Group 1 (sham-implanted and control tissues) resembled each other, with the few microscopic observations made being considered background. Placebo implantation was associated with implant-containing subconjunctival cavities, minimal-to-slight fibrous encapsulation, and a minimal-to-slight infiltrate of macrophages, sometimes as multinucleate cells, into the implant cavity (Table 3A and Fig. 3). The incidence and severity of the placebo-associated observations were dependent on neither the number nor the size of the devices. Latanoprost-containing implants were also associated with an implant-containing cavity, minimal fibrous encapsulation, and a minimal-to-slight infiltrate of macrophages into the implant cavity. Additional inflammatory cell infiltrates were often associated with drug-containing implants. Two males given 2 X50A devices containing latanoprost and 1 male and 1 female given 5 X190A devices containing latanoprost had a minimal or slight chronic–active inflammatory cell infiltrate into the tissue adjacent to the implants. The chronic–active inflammatory cell infiltrate was composed of a mixture of heterophils (neutrophils), macrophages, and lymphocytes. A minimal or slight infiltrate of heterophils was present within the implant cavity of 1 male and 3 females given 5 X190A devices containing latanoprost. The difference in the inflammatory cell infiltrates between the placebo- and drug-administered subconjunctival sites was considered latanoprost related, but not significant or adverse.
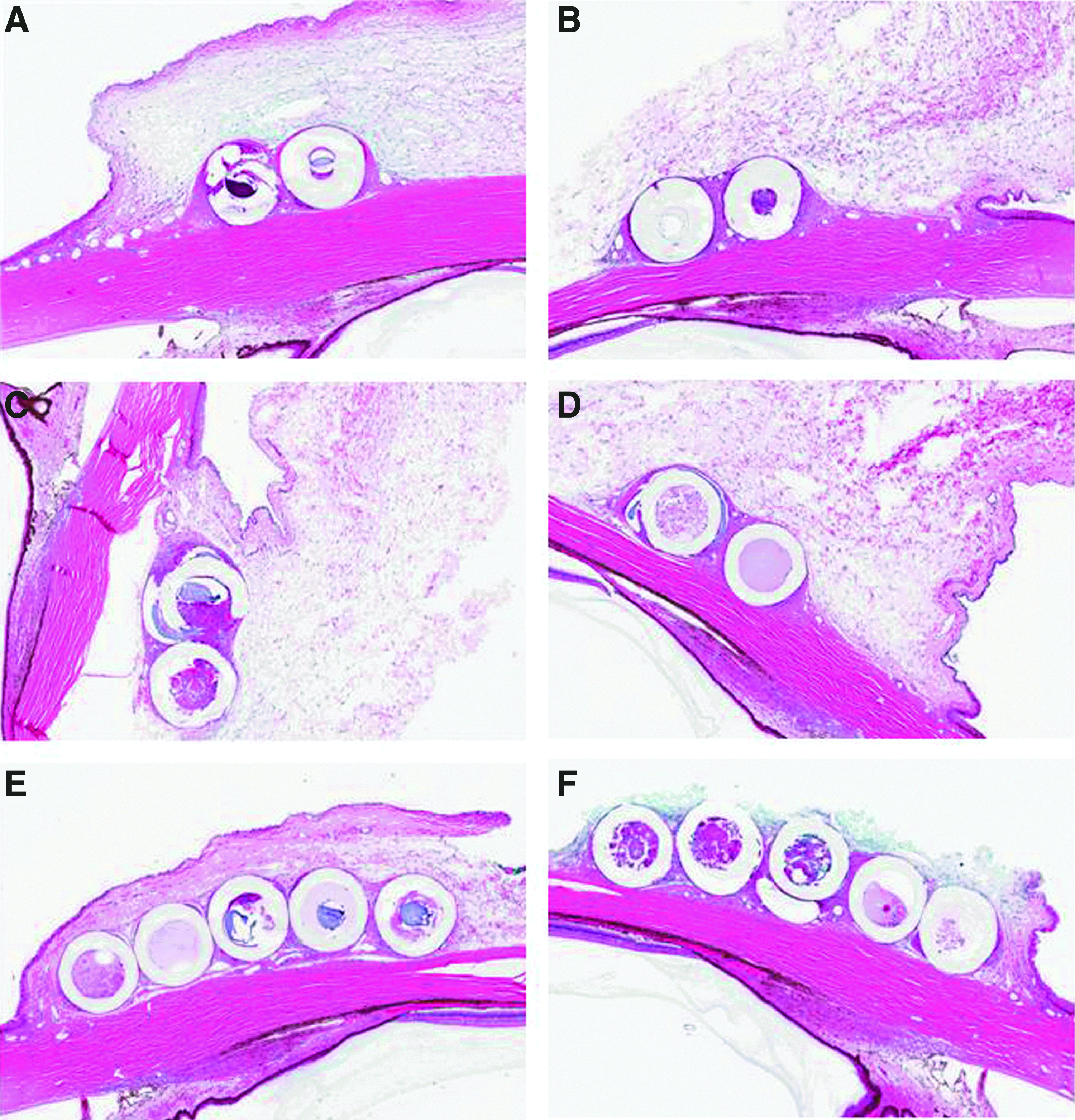
Three-month necropsy. Representative photographs at 5×magnification of ocular cross-sections containing placebo
None of the remaining microscopic observations was considered placebo- or test article-related, as they were considered common or background observations in this species and strain of the laboratory animal. Hypertrophy of retinal-pigmented epithelial cells in the peripapillary regions was common among all groups. This observation was considered a background observation seen in Dutch-belted rabbits and was independent of study-associated factors. 7 Additionally, some apparent differences in the inflammatory cell infiltrates were present in the eyelids (and eyelid conjunctiva), but these were considered insignificant background observations that differed among groups by chance.
Microscopic observations: 9-month (final) necropsy
Postimplantation, the left and right eyes from the control Group 1 sham-implanted and/or aseptically prepared eyes resembled each other. The few microscopic observations were considered background. Microscopic observations at the implant sites among 9-month animals at the scheduled necropsy included fibrous encapsulation and infiltrates of macrophages and lymphocytes around and within the cavities containing implants (Table 3B and Fig. 4). The observations at the placebo- and latanoprost-containing implant sites were similar. Heterophils were not common among the inflammatory infiltrates at the test article-containing implant sites. Except for the X50A devices, most of the implants were in varying degrees of disintegration and fragmentation, and there was ingrowth of fibrovascular tissue into the luminal remnant of the device. Occasionally, implant fragments appeared to have been engulfed by macrophages. The occasional fragmentation of the X50A devices may have been a result of the histological processing that included sectioning of the devices in situ. No difference in observations was noted among the placebo implants or those containing latanoprost. A minimal increase in encapsulation severity was present overall among animals receiving 5 implants, which seemed more related to the number of implants than to any inherent characteristic of the implants. None of the eyes in implanted groups had more severe or different adnexal, conjunctival, or eyelid microscopic observations compared with those of untreated or sham-implanted eyes. None of the intraocular observations were different or more severe among the groups of eyes. As at the 3-month necropsy, sham-implanted and control tissues resembled each other, and the few microscopic observations made were considered background. No histological findings were present in the remaining ocular tissues (data not shown).
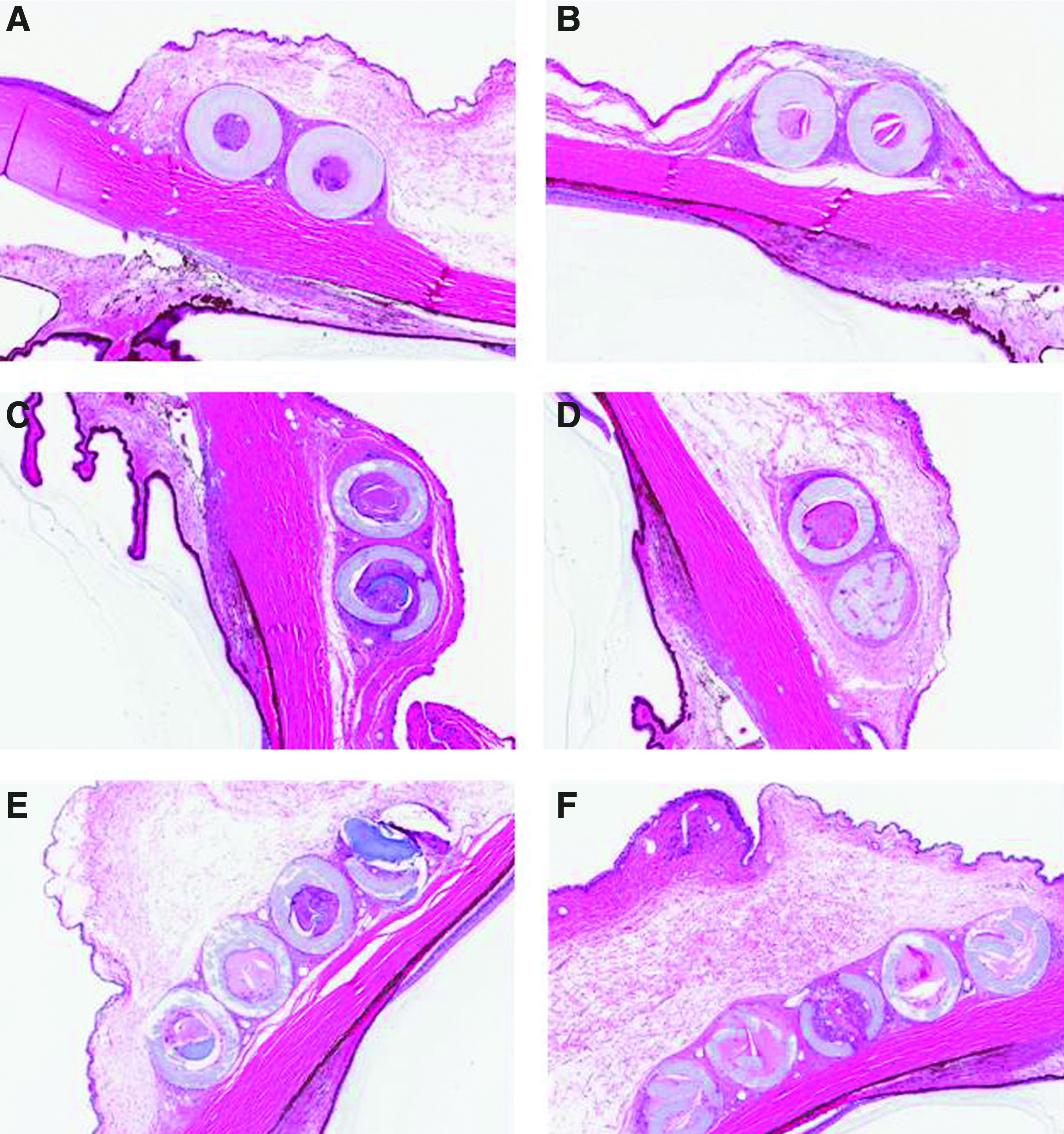
Nine-month necropsy. Representative photographs at 5×magnification ocular cross-sections containing placebo
Pharmacokinetics
Mean plasma TK parameters for latanoprost acid are summarized in Table 4. There were no quantifiable concentrations of latanoprost acid in samples analyzed from the control dose group animals (Group 1). Plasma latanoprost acid concentration–time data through to the end of the 9-month study showed no apparent sex-related differences in mean exposure. Therefore, data from male and female rabbits have been combined for purposes of reporting and discussion.
AUC(0–96), the area under the plasma latanoprost acid concentration–time curve.
The overall mean Tmax for latanoprost acid after subconjunctival placement of 2 X50A (Group 2), 2 X190A (Group 3), or 5 X190A (Group 4) devices containing latanoprost on study day 1 was observed at 1.0, 11, and 2.7 h, respectively (Table 3 and Fig. 5). The overall mean Cmax was 0.825, 1.60, and 3.04 ng/mL (Table 3 and Fig. 6), and the overall mean AUC(0–96) was 3.05, 29.2 and 68.3 ng·h/mL (Table 4 and Fig. 6) for Dose Groups 2, 3, and 4, respectively. The overall mean Cmax increased by 1.9-fold after an ∼3.8×increase in dose (2 X50A devices vs. 2 X190A devices). Similarly, the overall mean Cmax increased by 3.7-fold after an ∼9.5×increase in the dose (2 X50A devices vs. 5 X190A devices). The overall mean AUC(0–96) increased by 9.4-fold after an ∼3.8×increase in dose (2 X50A devices vs. 2 X190A devices). Similarly, the overall mean AUC(0–96) increased by 22-fold after an ∼9.5×increase in dose (2 X50A devices vs. 5 X190A devices). Therefore, the change in the overall mean Cmax was less than proportional to increases in the dose on study day 1. However, the change in systemic exposure, as measured by the overall mean AUC(0–96), was greater than proportional to increases in dose on study day 1.
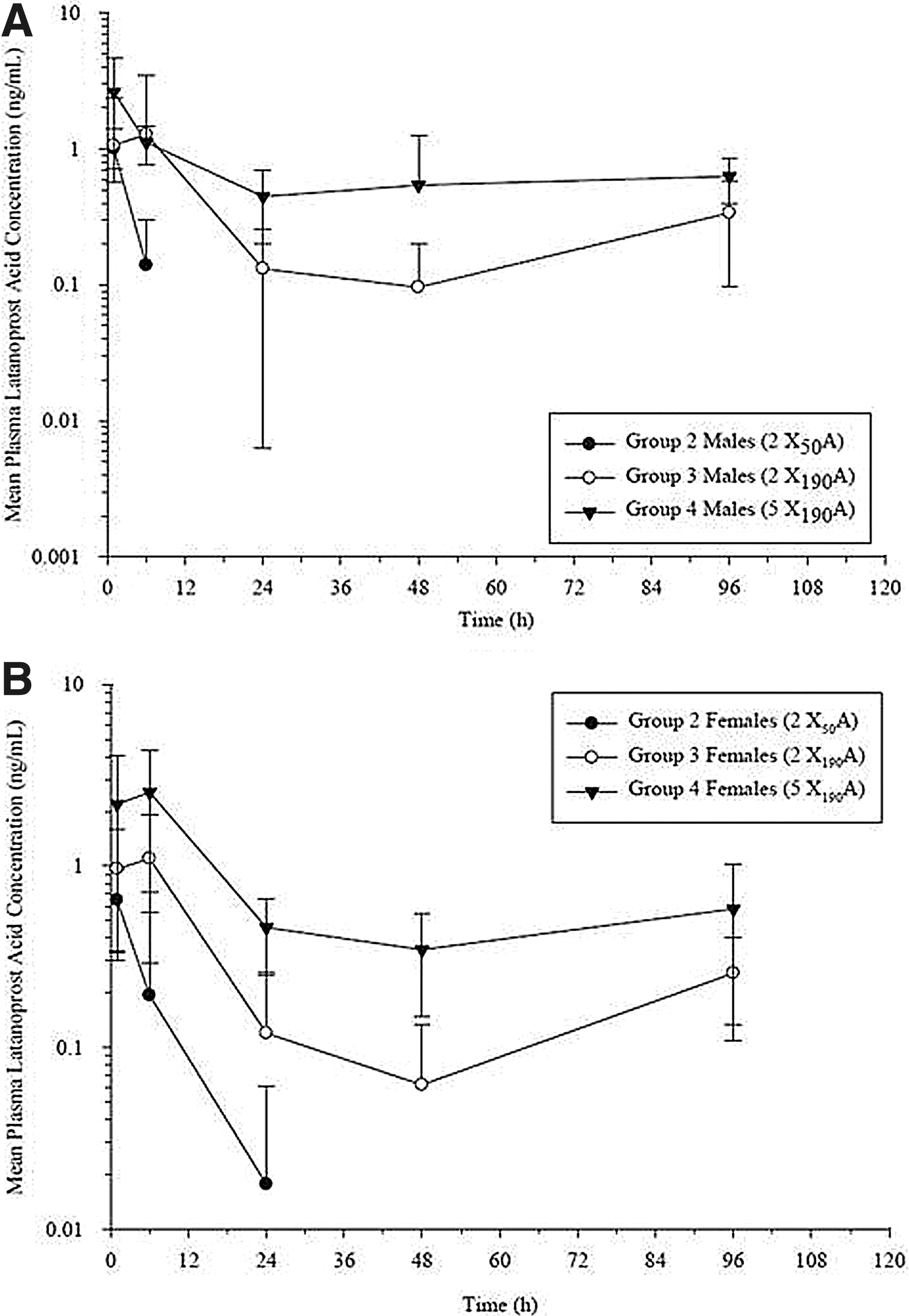
Mean (±SD) plasma latanoprost acid concentration in male
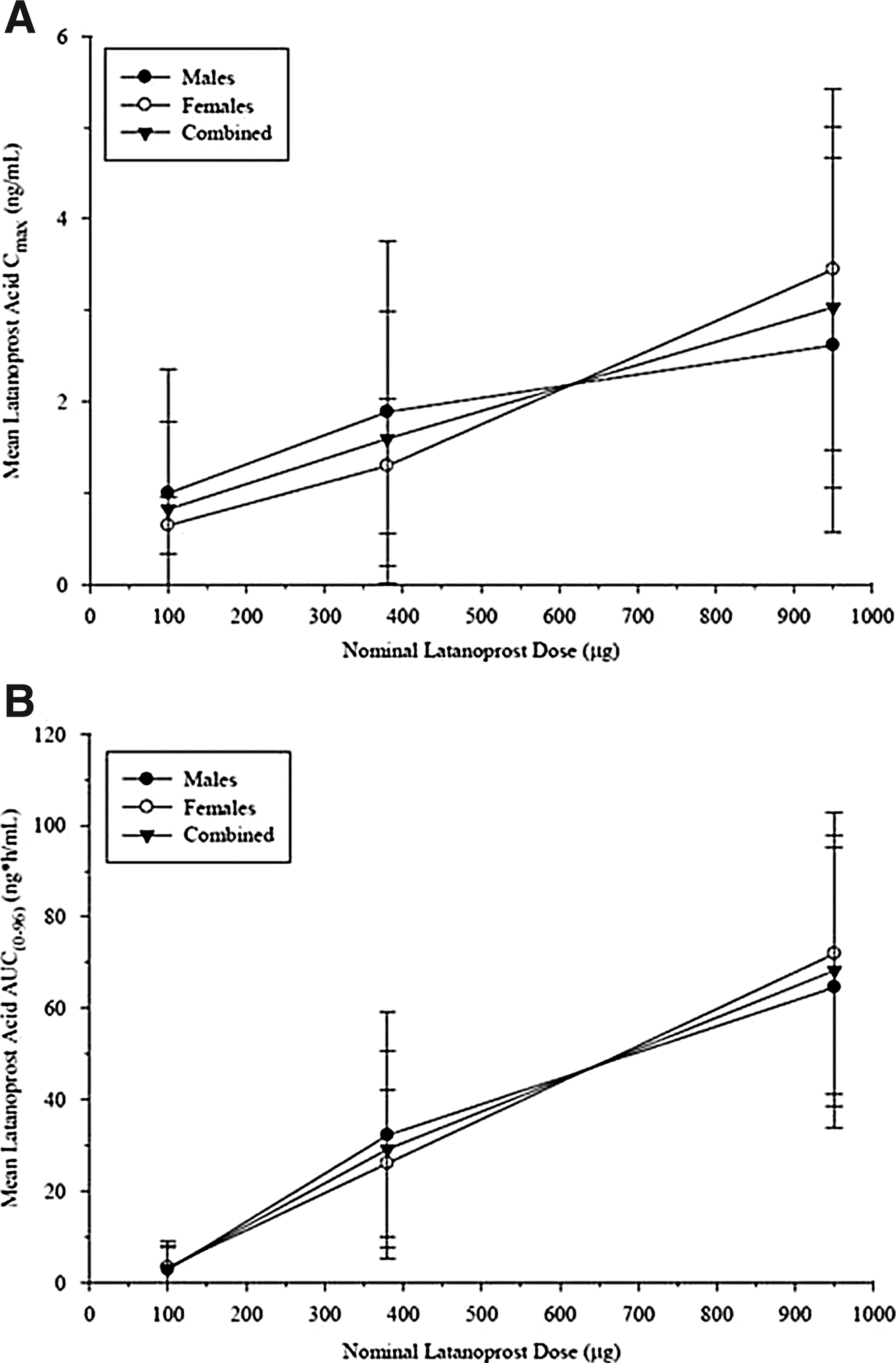
Mean (±SD) plasma latanoprost acid Cmax
The plasma concentration of latanoprost acid at 312 h postdose was below the limit of quantification (BLQ) for all animals, except 1 female in Group 2. The plasma latanoprost acid concentration was BLQ for 3 males and 1 female in Group 3 and BLQ for 1 male, but no females, in Group 4. The plasma latanoprost acid concentration was BLQ for animals in all dose groups at the week-14, week-25, and week-41 time points.
Discussion
The in vitro release rate profile observed with the latanoprost devices was similar to that described with biodegradable sustained release formulations of other glaucoma drugs. Hacker et al. studied the release characteristics of acetazolamide, dichlorphenamide, and timolol maleate in a fumarate-based drug delivery system. 8 In their study, the drug release profiles followed a 4-phase pattern. An initial burst phase in the first few days of testing followed by a long second phase (>100 days) during which a low, but consistent, release rate was observed. The third phase was characterized by a second burst that was longer in duration than the initial burst (∼100 days). The magnitude of this second burst was highly dependent on the loading concentration and physical chemical properties of the drug. While dicholphenamide and timolol maleate demonstrated secondary bursts of similar magnitude to their initial burst, acetazolamide had a very modest burst that was within 2-fold of the previous phase and was much shorter in duration. In the fourth phase, all drugs demonstrated a tapering-off in the release rate as the device fully degraded.
Like the findings with acetazolamide, dichlophenamide, and timolol maleate, the in vitro release profile of latanoprost device does demonstrate an initial burst followed by a low and near zero-order kinetic release rate for up to 90 days. Although the latanoprost devices were only characterized for the first 90 days in vitro, it is unlikely that they would have displayed a secondary burst, because the polymer shell of the device remains intact until complete depleting of the drug is achieved. Therefore, the release mechanism of the device continues to be the diffusion from the reservoir, followed by erosion of the polymer shell of the device subsequent to delivery of the entire contents.
The majority of findings in the 9-month study of latanoprost-containing devices were confined to the ocular surface as measured using the McDonald–Shadduck scoring method. The incidence, severity, and categories of findings reflect 3 phases within the study. In the first phase, represented by days 1 and 3, the findings appear to be dominated by the injection procedure itself (aseptic preparation of the eye, stabilization of the eye with forceps, and the injection itself) as evident by the similar severity and incidence of findings in the control groups relative to the groups implanted with devices. These handling procedures include administration of a general anesthetic, application of a sterilizing solution to the ocular surface, and physical manipulation of ocular tissues to allow or simulate injection of up to 5 devices in a single eye. In the next phase of the study represented by day 8 through week 9, the findings due to the injection procedure had resolved, and the findings in the implanted groups were limited to usually mild conjunctival congestion and swelling. The incidence and severity of these findings were dose-related, greater in latanoprost-containing devices relative to placebo devices, and lessened over the time period. Since conjunctival hyperemia is a known side effect of topical latanoprost treatment, its observation here would be consistent with an active drug being released up to 9 weeks in the study. Systemic exposure to latanoprost acid was detected at 2 weeks postimplantation, and while levels were undetectable at the next time point (14 weeks), low levels of drug may have been released locally in the eye, but did not reach detectable levels systemically. In the third phase, represented by weeks 13 through 39, there was an increase in the incidence of the minimal conjunctival congestion, but no difference between the placebo- and drug-treated eyes. The lack of a difference between the placebo- and drug-containing device eyes suggests that the latanoprost-containing devices were empty of their payload. The increased incidence in conjunctival congestion observed in this phase tended to diminish after the week-20 time point and may reflect an increase in inflammation due to the fragmentation of X190A devices that occurs as a part of the expected in situ disintegration of the device. This is supported by the observation that while the devices appear intact in histological sections from the day-93 necropsy, there was significant fragmentation of X190A devices observed in the histologic sections taken from the day-275 necropsy.
The ocular histopathologic changes were of the type and severity expected for a study with implantable devices of this size and composition. The presence of acute inflammation (heterophils) localized to the site of implantation is expected, especially during the early postimplantation phase. Macrophages, often present as multinucleate giant cells, were noted that had engulfed fragmenting debris from the devices. Heterophils and macrophages were limited to the subconjunctival areas immediately adjacent to the implant site. Topical ocular application of latanoprost is known to cause ocular and adnexal changes, such as eyelash lengthening, conjunctival hyperemia, and iridal darkening, 4 but inflammation is not typically seen with latanoprost. Therefore, the appearance of inflammatory cells is the most likely procedure- and device-related, and X190A device fragmentation probably contributed to this process.
As X190A devices fragmented, macrophages were seen infiltrating around devices as well as within luminal remnants of devices. The extent to which local release of inflammatory cellular enzymes and/or macrophage phagocytosis contributed to device fragmentation is not known. Compared to X190A devices, fracturing or fragmentation of X50A devices was less noticeable microscopically and, when present, was more likely due to sectioning or other histologic tissue-processing procedures. Fibrous encapsulation of devices was not considered overzealous and was of similar intensity across all devices. An appropriate, reparative fibrovascular response was noted near devices, consistent with a normal attempt at healing. Temporally, heterophilic inflammation generally decreased in affected tissues, such that by 9 months, there were few signs of acute inflammation.
After adjusting for the differences in body size between rabbits and humans (2.5 kg for rabbits versus 70 kg for human, a 28-fold difference), the Cmax resulting from the high-dose implant of ∼3 ng/mL is approximately twice the equivalent Cmax in humans following the recommended latanoprost clinical dose (53 pg/mL, a 56-fold difference). 9 However, the AUC(0–96h) of 68 ng h/mL resulting from the implants yields a much larger Cave than that in humans resulting from an AUC of 34 pg·h/mL following the latanoprost clinical dose. After adjusting for differences in body mass, the rabbit high-dose Cave was >17-fold that in humans. Therefore, the daily dose of latanoprost released from a single device is hypothetically in the range needed for the IOP-lowering efficacy based on both the in vitro release and rabbit systemic PK data. It should be noted that while the relationship between topical ocular dosing of latanoprost and the ocular and systemic exposure to latanoprost acid has been well established, the novel route of dosing and lack of ocular exposure measurements in this study make conclusions of efficacious exposure speculative.
It should also be pointed out that while the rabbit is a well-established ocular safety model and was used on the nonclinical toxicology evaluation of latanoprost, supporting its initial marketing application, it has certain limitations. For example, rabbits are known to lack the IOP-lowering response to latanoprost. 10 Further, the increased iridal pigmentation noted in labeling information for latanoprost has not been observed in other species, such as the rabbit (Pfizer internal data not shown). Further, since the main goal of this study was to evaluate safety, it is limited in its ability to provide a nonclinical proof of concept for the therapeutic value of the sustained delivery system. To accomplish this, studies incorporating ocular pharmacokinetics and pharmacodynamics (IOP) in a latanoprost-sensitive model are required. However, the goal of this study was to evaluate the safety of the sustained latanoprost delivery system, and not the safety and efficacy of latanoprost, which have been established elsewhere.
The data reported here demonstrate that a subconjunctivally implanted device made of either a polyimide or poly-lactic-co-glycolic acid material and delivering latanoprost over an extended period is well tolerated in rabbits for up to 9 months. The effects related to the implantation procedure will be minimized in humans by implanting a single device per eye, dosing a single patient per visit, using a local, but not general, anesthetic, and using a less-rigorous sterilization procedure. Further, the in vitro characteristics and extrapolated systemic exposure achieved with the X190A biodegradable device are consistent with reaching efficacious ocular levels in humans. Therefore, the positive results obtained here warrant further exploration of the use of this delivery system for the sustained treatment of glaucoma.
Footnotes
Acknowledgments
This study was conducted at the Covance Laboratories and supported by funding from Pfizer, Inc. Work by pSivida employees was performed as a part of a contractual collaborative agreement with Pfizer, Inc.
Author Disclosure Statement
No competing financial interests exist.