
Abstract
Abstract
Purpose:
To investigate the underlying mechanism by which pirfenidone blocks the transition from the G1 to S phase in primary human Tenon's fibroblasts.
Methods:
Primary human Tenon's fibroblasts were characterized by immunocytofluorescence staining with vimentin, fibroblast surface protein, and cytokeratin. After treating Tenon's fibroblasts with pirfenidone under proliferation conditions (10% fetal bovine serum), cell proliferation was measured using a WST-1 assay. Progression through the cell cycle was analyzed by flow cytometry. The expression of CDK2, CDK6, cyclinD1, cyclinD3, and cyclinE and the phosphorylation of AKT, ERK1/2/MAPK, JNK/MAPK, and p38 MAPK were estimated using western blot analysis.
Results:
Under proliferative conditions, pirfenidone inhibited Tenon's fibroblasts proliferation and arrested the cell cycle at the G1 phase; decreased the phosphorylation of AKT, GSK3β, ERK1/2/MAPK, and JNK/MAPK; increased the phosphorylation of p38 MAPK; and inhibited CDK2, CDK6, cyclin D1, cyclin D3, and cyclin E in a dose-dependent manner. Inhibitors of AKT (LY294002), ERK1/2 (U0126), and JNK (SP600125) arrested the G1/S transition, similar to the effect of pirfenidone. The p38 inhibitor (SB202190) decreased the G1-blocking effect of pirfenidone. The expression of CDK2, CDK6, cyclin D1, and cyclin D3 were inhibited by LY294002, U0126, and SP600125. SB202190 attenuated the pirfenidone-induced reduction of CDK2, CDK6, cyclin D1, cyclin D3, and cyclin E.
Conclusions:
Pirfenidone inhibited HTFs proliferation and induced G1 arrest by downregulating CDKs and cyclins involving the AKT/GSK3β and MAPK signaling pathways.
Introduction
G
Pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone, PFD) is a novel agent that shows antifibrotic, anti-inflammatory, and antioxidative effects 13 in animal experiments and clinical trials in pulmonary, liver, cardiac, and renal fibrosis.14–18 Increased bleb survival rate and decreased toxicity has also been observed in a glaucoma filtering surgery model with PFD. 19 Its antifibrotic action is primarily attributed to its antagonism of fibroblast proliferation and migration and the reduction of extracellular matrix deposits. 20
We previously found that PFD inhibited Tenon's fibroblast proliferation and arrests the fibroblast cell cycle in the G1 phase. 21 However, the mechanism by which PFD regulates the cell cycle in human Tenon's fibroblasts (HTFs) is poorly understood. The transition from G1 to S phase is primarily regulated by the activation of the cyclin-dependent kinase 2 (CDK2)/cyclin E and CDK4/6/cyclin D complex, leading to the downstream phosphorylation of the retinoblastoma protein (Rb) and relieving the inhibitory effect of Rb on E2F to accelerate the transition.22,23
Many signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, 24 extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK)/MAPK, 25 and p38 MAPK pathways, participate in the fibrosis process and impact fibroblast proliferation. 26 A crucial downstream protein in the AKT pathway is GSK3β, which can phosphorylate cyclin D, leading to its rapid ubiquitination and proteasomal degradation of CDKs. It then inhibits the G1 to S transition. 27 The ability of PFD to inhibit Tenon's fibroblast proliferation and arrest the cell cycle through these signaling pathways and the roles of these pathways in regulating the CDK/cyclin complex in Tenon's fibroblasts have not previously been investigated.
In this study, we found that PFD can arrest the cell cycle by downregulating CDK2, CDK6, cyclin D, and cyclin E expression. The AKT/GSK3β, ERK/MAPK, JNK/MAPK, and p38 MAPK pathways were found to be involved in the downregulation of these cell cycle-regulated proteins.
Methods
Cell culture and treatment
Human Tenon's capsule samples were obtained from 9 selected patients during strabismus surgery after signed informed consent was obtained. We cultured cells from these 9 donors at line 1, then mixed these cells and continue to culture them until line 5. Line 3 to line 5 of these mixed cells were chosen for all experiments. All procedures involving human tissues were performed according to the tenets of the Declaration of Helsinki and with approval from the Institutional Review Board of Sun Yat-sen University in Guangzhou.
Tenon's capsules were dissected into 2 × 2 mm sections and placed in 25 cm2 culture bottles in Dulbecco's modified Eagle's medium (DMEM; Gibco-Life-Technology) supplemented with 10% fetal bovine serum (FBS; Gibco- Life-Technology). Cells were migrated from explants and then incubated at 37°C in a 5% CO2 and humidified atmosphere. We adopted serum-activated fibroblasts as proliferative cell model in this study.
Immunocytofluorescence
HTFs were seeded in 6-well plates at a concentration of 1 × 105 cells/well and incubated in complete culture media overnight at 37°C to allow the cells to adhere. The culture media was then removed. The next procedures were performed as described previously. 21 Images were observed and photographed using a laser scanning confocal microscopy (LSM510META; Carl Zeiss).
Cell proliferation assay
Cell proliferation was measured using a water-soluble tetrazolium salt (WST)-1 assay kit (Roche) according to the manufacturer's instructions. HTFs were seeded in 96-well plates at a concentration of 3,000 cells/well with complete culture media for 12 h and then incubated in FBS-free medium for 24 h for synchronization. FBS-free medium, 10% FBS and different concentrations of PFD (0, 0.1, 0.3, 0.6, 0.9, and 1.2 mg/mL) (Sigma-Aldrich) with 10%FBS were applied to the wells for 0, 12, 24, and 48 h at 37°C. After these treatments, WST-1 (10 μL) was added to each well. The plates were incubated for another 1.5 h at 37°C. After incubation, absorbance was measured at 450 nm (ELx800; BioTek).
Cell cycle analysis
Into each well of a 6-well tissue culture plate, 1 × 105 HTFs were placed in 10% FBS-DMEM and incubated for 12 h. Cells were then cultured in FBS-free DMEM for 24 h. They were then incubated in FBS-free DMEM containing the AKT inhibitor LY294002 (20 μM; CST), ERK inhibitor U0126 (20 μM; CST), JNK inhibitor (SP600126 20 μM; CST), or p38 inhibitor SB 292109 (20 μM; CST) for 2 h. All inhibitors were prepared in DMSO as stock solutions (20 mM), stored at −20°C. After treatment with the inhibitors, the cell culture medium was replaced with 10% FBS-DMEM and incubated for another 24 h in the absence or presence of PFD.
After the various treatments, the cells were harvested with 0.25% trypsin, washed in PBS, and fixed with 70% ethanol at 4°C overnight. Fixed cells were treated with 50ug/mL RNase (Wako Pure Chemicals) at 37°C for 30 min, then subjected to PI/RNase staining followed by flow cytometry analysis using a cytometer (BD FACS Calibur; BD Biosciences), and the cycle distribution was analyzed with commercial software (Cell Quest Pro; BD Biosciences).
Preparation of protein lysates and western blot analysis
Western blot analysis has been done after 24 h treatment with PFD. Cells were rinsed 3 times with PBS and harvested in RIPA buffer (CST) containing a protease inhibitor cocktail (Roche) and a phosphatase inhibitor (Roche) according to the manufacturer's instructions. After incubation on ice for 5 min, the lysates were centrifuged at 12,000 g for 5 min. The supernatant proteins were quantified using a Nanodrop2000 (Thermo Scientific). An equal amount (30 μg) of each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride membranes (PVDF; Millipore).
After blocking with 5% BSA in Tris-buffered saline containing 0.1% Tween-20 (TBST) at room temperature for 2 h, blots were incubated with anti-anti-CDK2 (CST; 1:1,000 dilution), anti-CDK6 (CST; 1:1,000 dilution), anti-cyclin D1 (CST; 1:1,000 dilution), anti-cyclin D3 (CST; 1:1,000 dilution), anti-cyclin E (CST; 1:1,000 dilution), anti-AKT (CST; 1:1,000 dilution), anti-phospho-Akt (Thr308) (CST; 1:1,000 dilution), anti-phospho-Akt (Ser473) (CST; 1:1,000 dilution), anti-ERK1/2 (CST; 1:1,000 dilution), anti-phospho-ERK1/2 (CST; 1:1,000 dilution), anti-JNK/SPAK (CST; 1:1,000 dilution), anti-phospho-JNK/SPAK (CST; 1:1,000 dilution), anti-P38/MAPK (CST; 1:1,000 dilution), anti-phospho-P38/MAPK, anti-GAPDH (CST; 1:2,000 dilution), and anti-β-actin (CST; 1:2,000 dilution) primary antibodies at 4°C overnight.
Blots were washed with TBST and then incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit IgG (CST; 1:2,000 dilution) for 1 h at room temperature. Membranes were exposed with an ECL system (Millipore Corp.) and quantified by densitometry, correcting for the background density of each gel. β-actin and GAPDH were used as loading controls.
Statistics analysis
All experiments were repeated at least 3 times. The results are expressed as the mean ± SD. Different groups were assessed using a one-way analysis of variance (ANOVA) and Tukey's post hoc test. A comparison between 2 groups was compared by a 2-tail Student's t test. P < 0.05 indicated statistically significant differences.
Results
HTFs characterization
Immunofluorescence staining was used to identify fibroblasts (Fig. 1). The vimentin staining showed red fluorescence in the cytoplasm with oval-shaped, blue-stained nuclei (Fig. 1A). The FSP antibody staining showed green fluorescence in the cytoplasm with blue nuclei (Fig. 1B). The cytokeratin staining was negative (green if positive) with blue nuclei (Fig. 1C). Therefore, the cultured cells proved to be fibroblasts rather than other cells at ocular surface such as conjunctival or corneal epithelium.
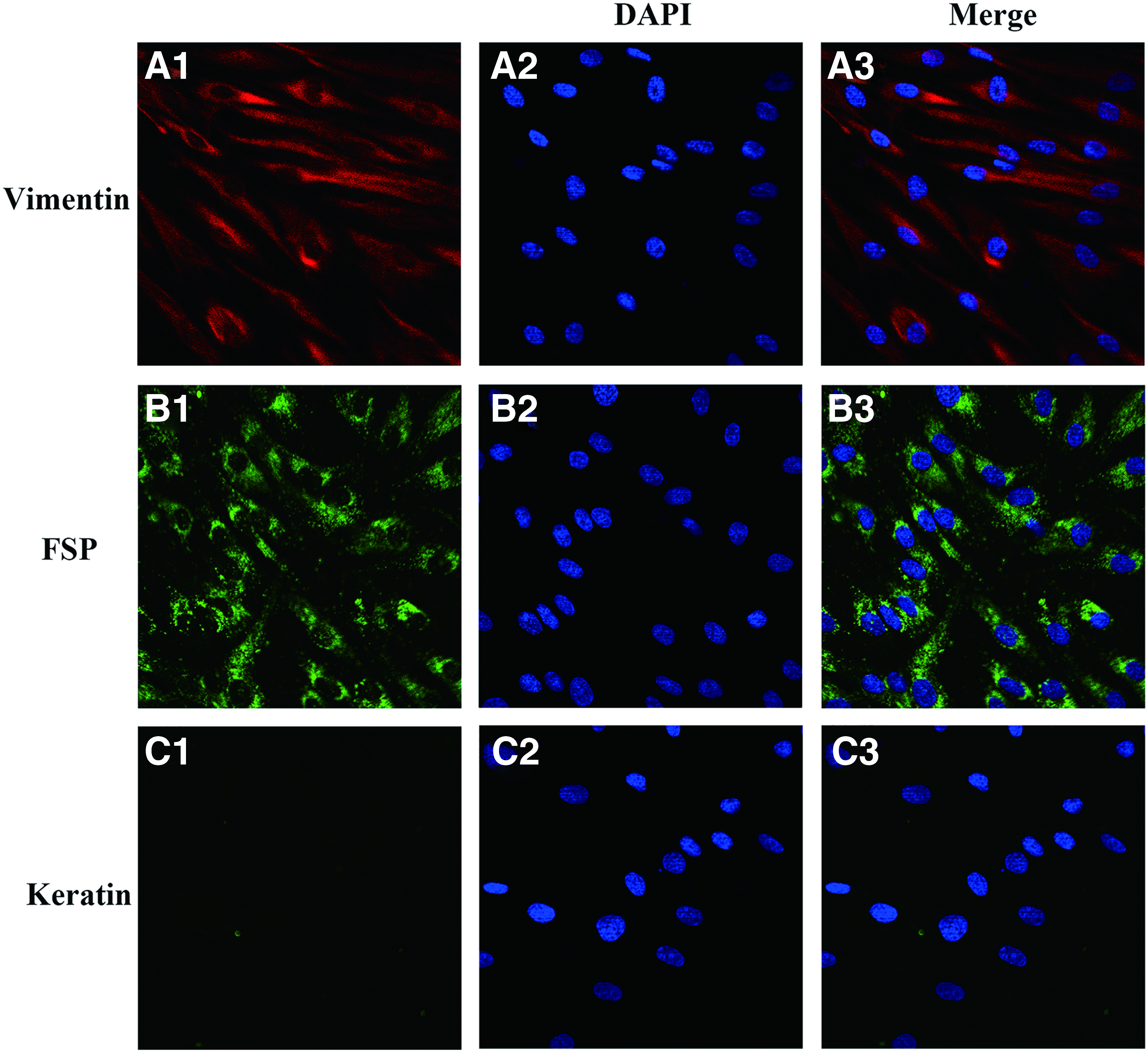
Characterization of HTFs. HTFs incubated with anti-vimentin
PFD inhibited HTFs proliferation and induced G1 arrest
The HTFs were incubated in 10% FBS-DMEM containing PFD at concentrations of 0, 0.1, 0.3, 0.6, 0.9, and 1.2 mg/mL for 12, 24, and 48 h. The HTFs were cultured in 10% FBS-DMED or serum-free DMEM as a control. As demonstrated by the WST-1 assay results (Fig. 2), 10% FBS significantly stimulated the proliferation of HTFs. PFD inhibited HTF proliferation in a dose- and time-dependent manner.

PFD inhibited HTFs proliferation and arrested the cell cycle at G1 phase. Three independent experiments were conducted. The WST-1 assay revealed that HTF proliferation was inhibited by PFD in a dose- and time-dependent manner. The results are presented as the mean ± SD, **P < 0.01 versus Control, comparison between the cells with PFD exposure and the control cells with 10% FBS at different time points
At 12 h, PFD concentrations of 0.6 mg/mL or greater significantly inhibited HTFs proliferation compared with 10% FBS-DMED group, significant difference among 0.6, 0.9, and 1.2 mg/mL group (P < 0.01); at 24 h, PFD concentrations of 0.3 mg/mL or greater significantly inhibited HTFs proliferation (P < 0.01), no significant difference among 0.6 mg/mL and greater concentrations; at 48 h, all concentrations of PFD inhibited HTFs proliferation (P < 0.01), no significant difference among 0.6 mg/mL and greater concentrations (Fig. 2A).
The cell cycle analysis demonstrated that the number of HTFs in the G1 phase increased, the S and G2/M phase decreased in a dose-dependent manner (P < 0.05) (Fig. 2B, C).
PFD induced HTFs G1 arrest through the inhibition of the AKT/GSK3β, ERK/MAPK, and JNK/MAPK signaling pathways
Phosphorylation at a threonine (Thr308) and serine residue (Ser473) is essential for the activation of AKT. Phospho-AKT can further phosphorylate GSK3β, an inhibitor of cyclin D, at serine residue 9 (Ser9), which leads to the inactivation of GSK3β. PFD can suppress the phosphorylation of AKT at Thr308 and Ser473 to further reduce GSK3β inactivation (Fig. 3A). Activation of ERK1/2/MAPK and JNK/MAPK by phosphorylation can promote fibroblast proliferation and the transition from the G1 to S phase. Phosphorylation of ERK1/2/MAPK and JNK/MAPK can be downregulated by PFD (Fig. 3A). The inhibitors of AKT (LY294002), ERK1/2 (U0126), and JNK/MAPK (SP600125) induced G1 arrest in the HTFs (Fig. 3B), suggesting that PFD induces HTFs G1 arrest by inhibiting the AKT/GSK3β, ERK/MAPK, and JNK/MAPK signaling pathways.
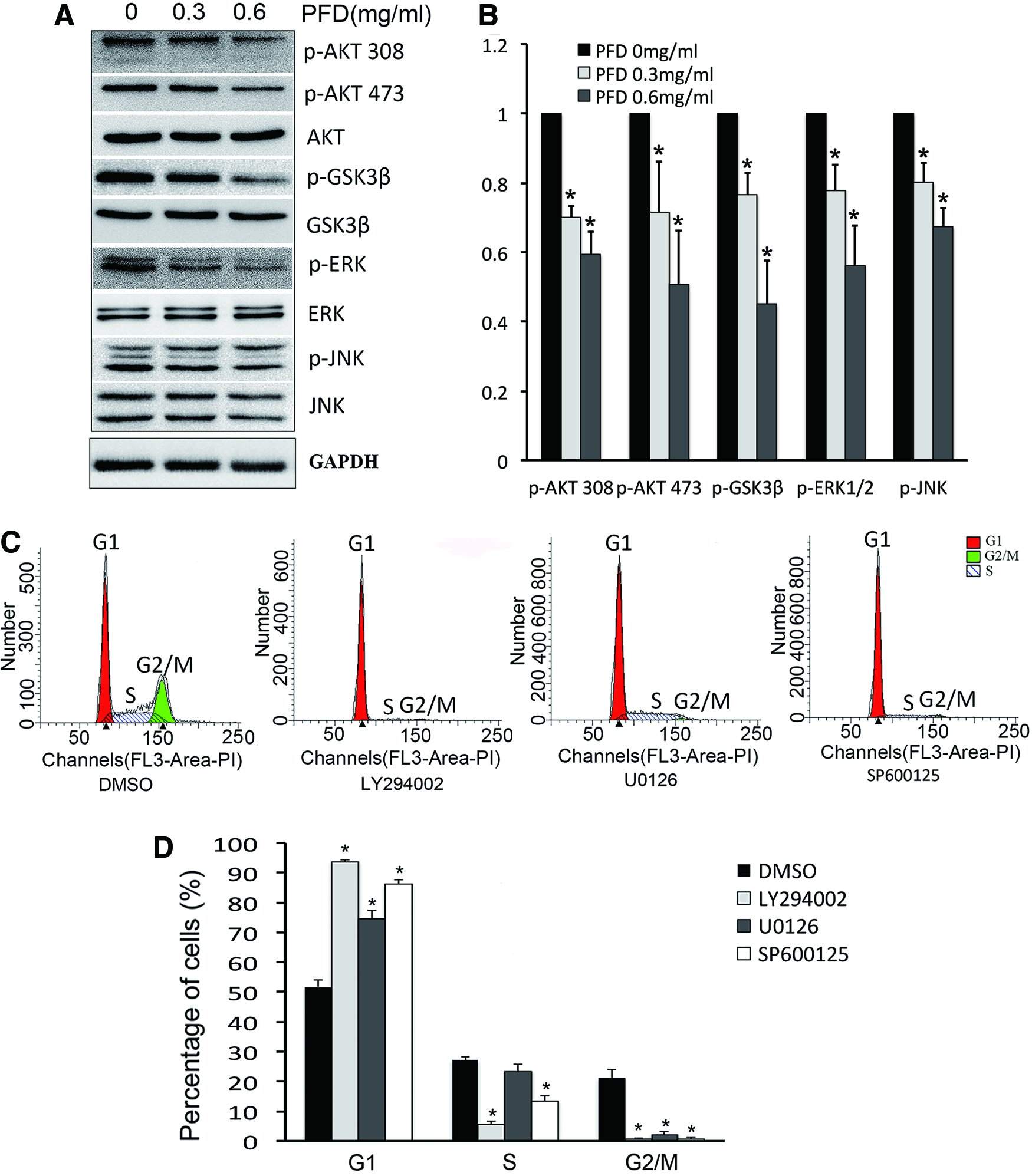
PFD induced HTF G1 arrest by inhibiting the AKT/GSK3β, ERK/MAPK, and JNK/MAPK signaling pathways. Western blot has been done after 24 h treatment with PFD. Western blot analysis revealed that phosphorylation of AKT, GSK3β, ERK1/2/MAPK, and JNK/MAPK was downregulated by PFD in a dose-dependent manner. The band densities of phosphorylated AKT, GSK3β, ERK1/2, and JNK were normalized to their corresponding total protein levels.
PFD induced HTFs G1 arrest through activation of the p38 MAPK signaling pathway
Various antineoplastic chemicals revealed antiproliferative mechanisms involving the activation of the p38 MAPK pathway that induced cell cycle arrest. PFD can increase the phosphorylation of p38 MAPK and, consequently, can activate it (Fig. 4A). The specific inhibitor SB202190 accelerated the transition from the G1 to S phase (Fig. 4B), which confirmed the effect of p38 MAPK-induced G1 arrest. The G1 arrest induced by PFD can be partly decreased by SB202190, which suggests that PFD induces HTF G1 arrest by activating the p38 MAPK signaling pathway (Fig. 4B).
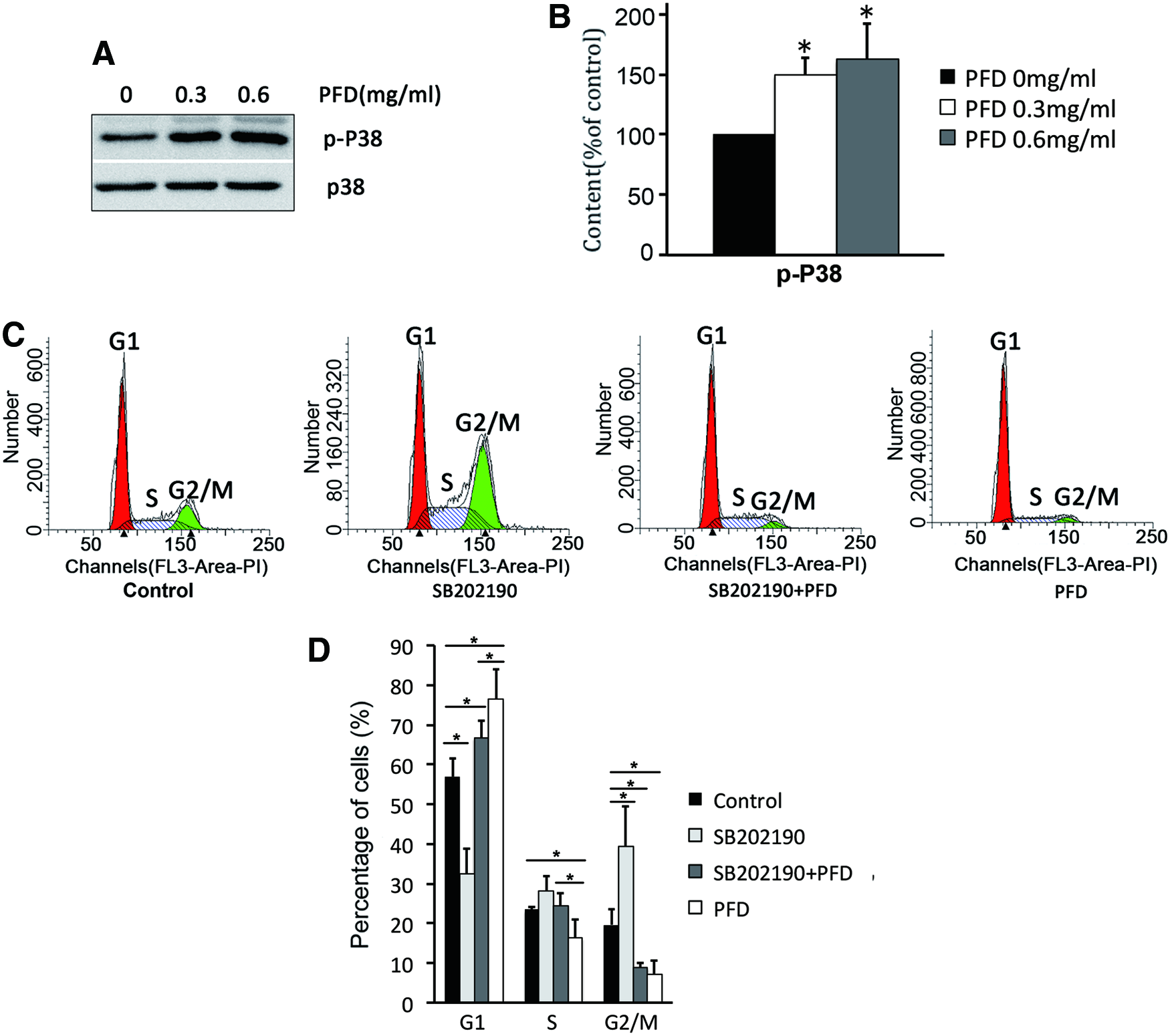
PFD induced HTFs G1 arrest by activating the p38 MAPK signaling pathway. Cells were incubated in serum-free DMEM containing the p38 inhibitor SB 292109 for 2 h. Then, the cell culture medium was replaced with 10% FBS-DMEM and incubated for another 24 h in the absence or presence of PFD. PFD increased the phosphorylation of p38 by western blot
PFD arrested the G1 phase by downregulation of CDK6/cyclin D and CDK2/cyclin E
PFD arrested the cell cycle, which was confirmed by the reduced expression of the proteins that accelerate the transition from G1 to S phase, including CDK2, CDK6, cyclin D1, cyclin D3, and cyclin E (Fig. 5). These results indicate that PFD inhibits HTFs proliferation and arrests the cell cycle by downregulating the CDK6/cyclin D and CDK2/cyclin E complexes.
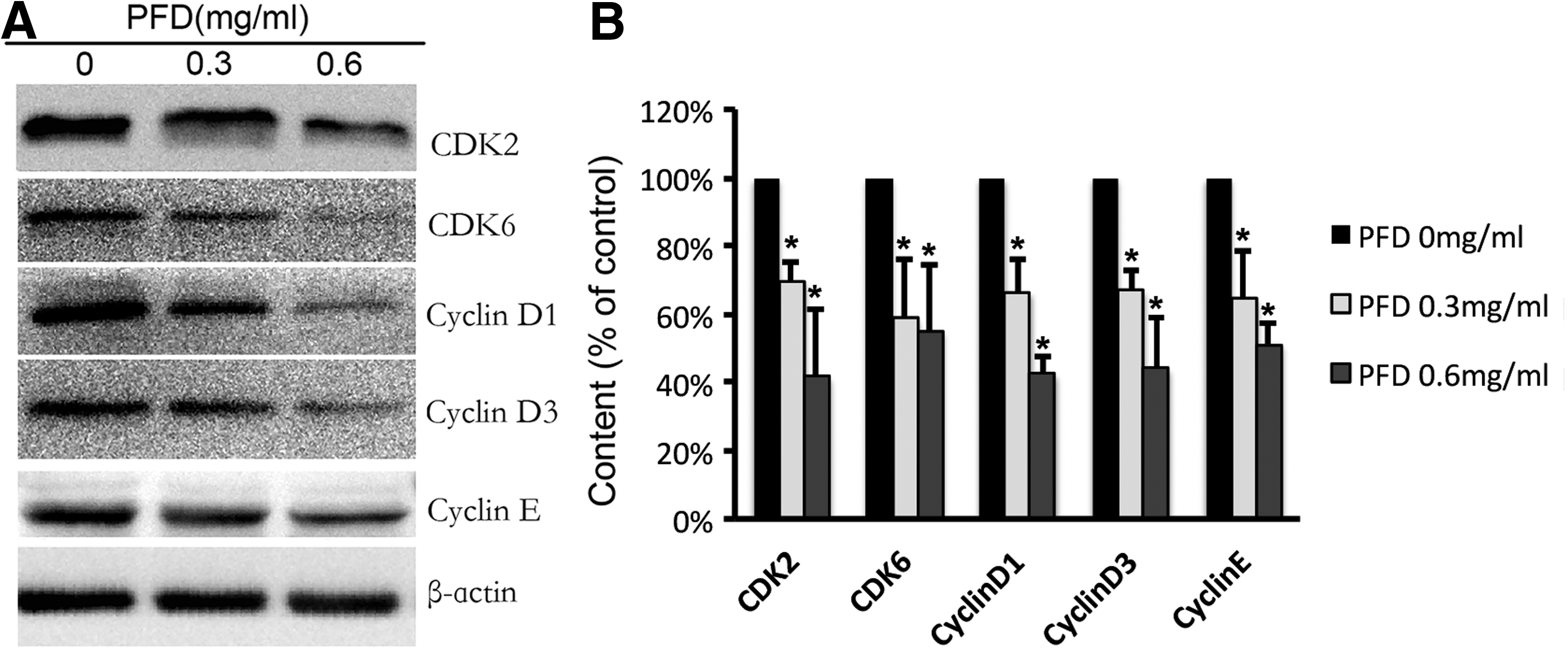
The expression of CDK2, CDK6, cyclin D1, cyclin D3, and cyclin E was inhibited by PFD
PFD downregulated CDK2, CDK6, and cyclin D proteins involved in the suppression of AKT, ERK/MAPK, and JNK/MAPK signals
The AKT inhibitor LY294002 suppressed the expression of CDK2, CDK6, cyclin D1, and cyclin D3 (Fig. 6). The ERK/MAPK inhibitor U0126 also suppressed the expression of CDK2, CDK6, and cyclin D1 (Fig. 6). The JNK/MAPK inhibitor SP600125 downregulated CDK2, CDK6, cyclin D1, and cyclin D3 (Fig. 6). No tested inhibitor affected the expression of cyclin E. The above results suggest that PFD downregulated CDK2, CDK6, and cyclin D proteins involved in the inactivation of AKT, ERK/MAPK, and JNK/MAPK signals.
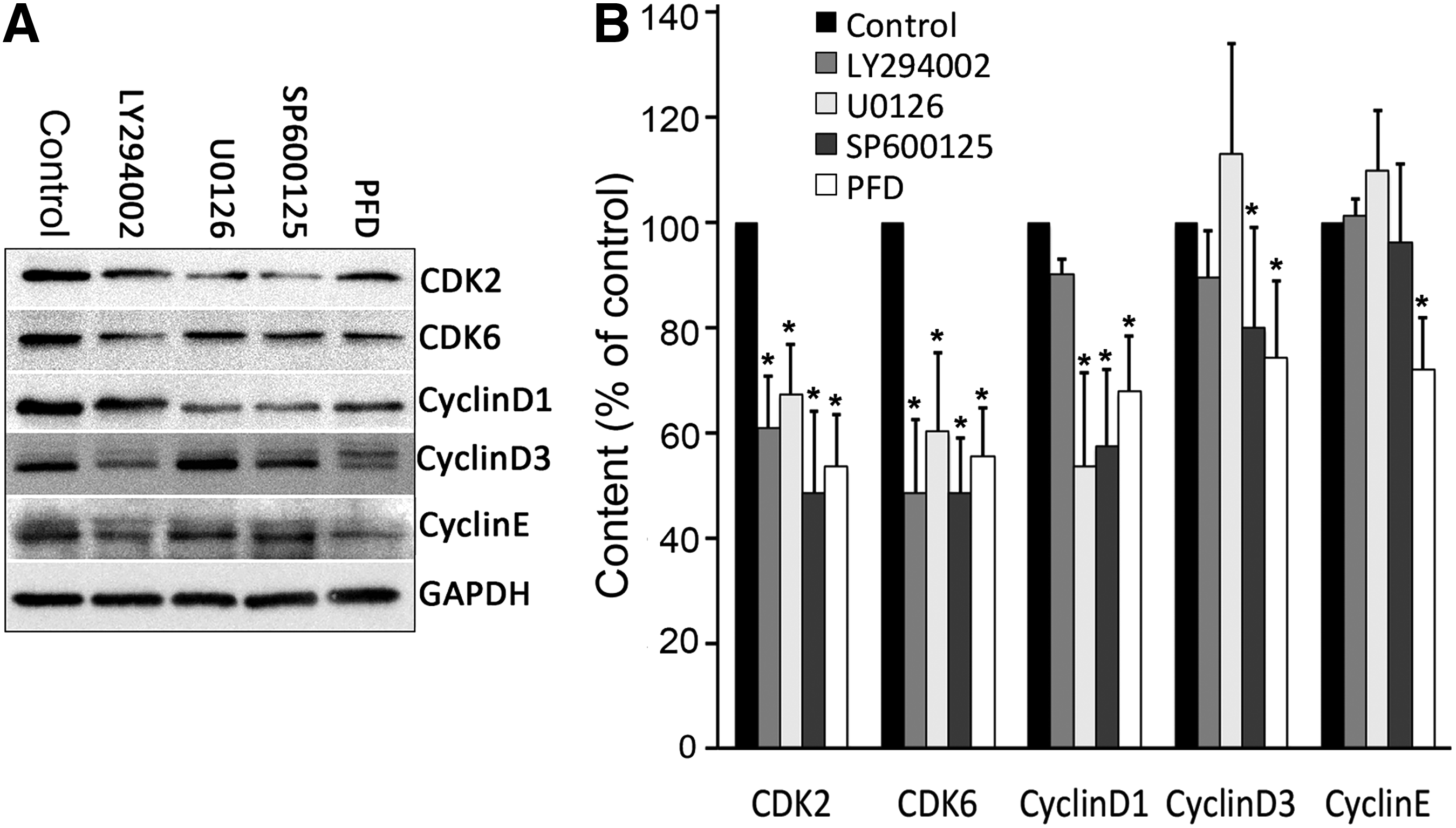
Western blot analysis showed that PFD downregulated CDK2, CDK6, and cyclin D proteins that are involved in the suppression of AKT, ERK/MAPK, and JNK/MAPK signals. Cells were incubated in serum-free DMEM containing the following inhibitors for 2 h. Then the cell culture medium was replaced with 10% FBS-DMEM and incubated for another 24 h in the absence or presence of PFD. The AKT inhibitor LY294002 suppressed the expression of CDK2, CDK6 and cyclin D1 and cyclin D3
PFD downregulated CDK2, CDK6, cyclin D, and cyclin E proteins involved in the activation of the p38 MAPK signal
The p38 MAPK inhibitor SB202190 significantly increased the expression of CDK2, cyclin D1, cyclin D3, and cyclin E (Fig. 7). PFD inhibited the expression of CDK2, CDK6, cyclin D1, and cyclin E, which was reversed by SB202190. This implied that PFD suppressed the expression of CDK2, CDK6, cyclin D, and cyclin E proteins involving in the activation of p38/MAPK signal (Fig. 7).
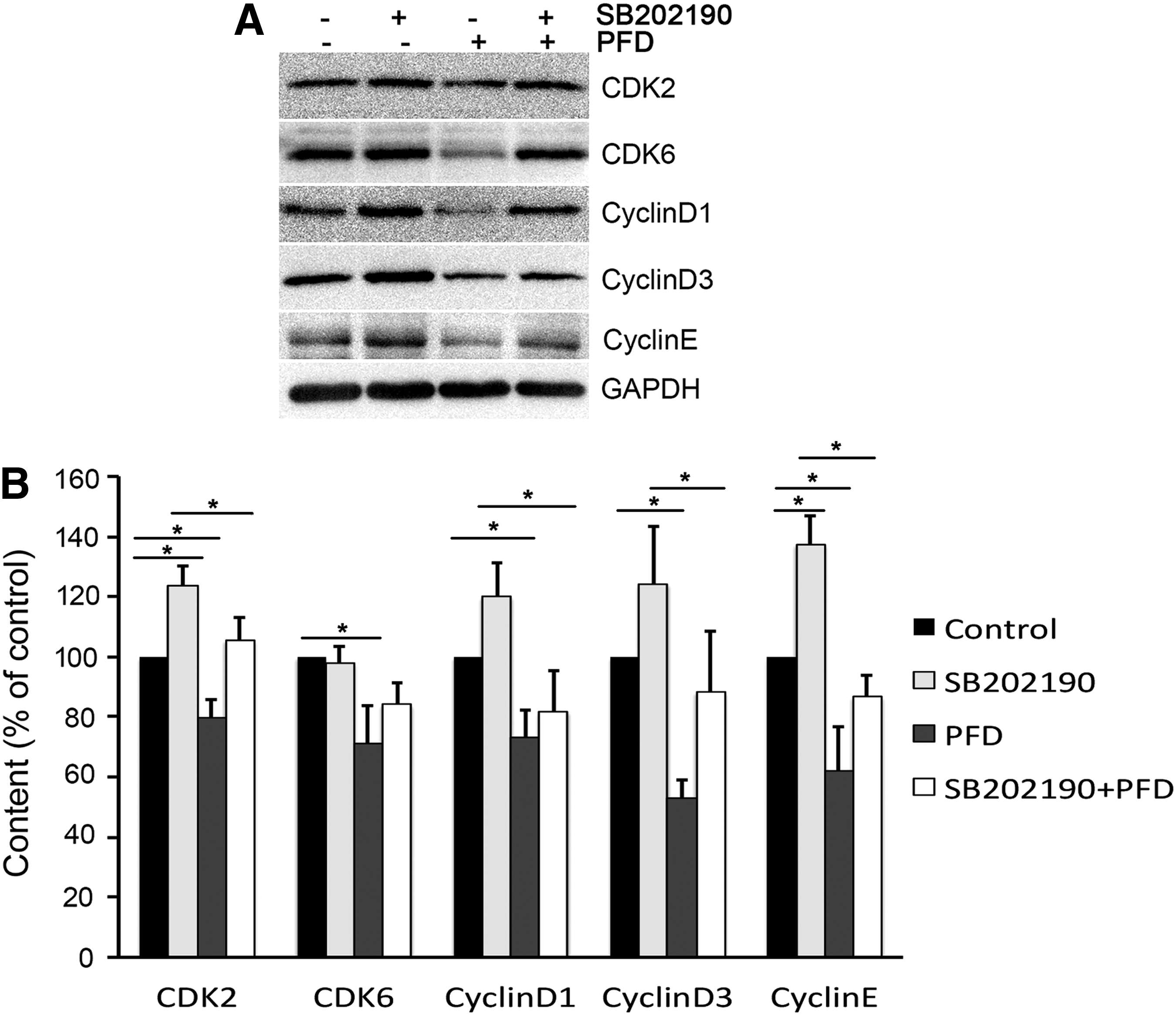
PFD downregulated CDK2, CDK6, cyclin D, and cyclin E proteins involved pn the activation of the p38 MAPK signal by western blot. Cells were incubated in the p38 inhibitor SB 292109 for 2 h, and then cultured for another 24 h in 10% FBS-DMEM in the absence or presence of PFD. The p38 MAPK inhibitor SB202190 significantly increased the expression of CDK2, cyclin D1, cyclin D3, and cyclin E. PFD inhibited the expression of CDK2, CDK6, cyclin D1, and cyclin E, which was reversed by SB202190
Discussion
Fibrosis at the surgery site after glaucoma filtering surgery is a natural wound healing process. Fibroblast proliferation plays an indispensable role in subconjunctival scarring. Downregulation of CDKs and cyclins inhibits the progression of the cell cycle. In this study, PFD intensively inhibited HTFs proliferation and led to G1 arrest by suppressing the expression of CDK2, CDK6, cyclin D, and cylin E, kinases that accelerate the transition from the G1 to S phase.
PFD inhibits pulmonary fibroblast proliferation, a process that involves the AKT pathway, 28 and suppresses the secretion of hyaluronic acid synthase by orbital fibroblasts, which involves both AKT and ERK1/2 signals. 29 However, no study has investigated which pathway involved in cell cycle regulation in HTFs is inhibited by PFD. Our results first revealed that PFD suppressed the activation of AKT and ERK1/2 in HTFs and increased the activation of GSK3β, which is the downstream signal of AKT. The inhibitors of AKT and ERK1/2 demonstrated a similar cell cycle arrest effect and downregulated CDK2, CDK6, and cyclin D. Feng et al. demonstrated that fluorofenidone, another pyridone agent, decreased liver fibrosis and suppressed hepatic stellate cell proliferation via a JNK/MAPK signal. 30 Our study first revealed that PFD induced G1 arrest via the suppression of the JNK/MAPK signal.
Previous studies have implied that p38 MAPK negatively regulate the cell cycle transition and induce G1 or G2/M arrest. 31 However, some results have also revealed positive effects of p38 MAPK on cell cycle progression. 32 The role of p38 MAPK in the regulation of the cell cycle appears to be dependent on the stimulus and cell types in different pathologic processes. 33 The suppression of the G1-S transition by p38 MAPK was related to the stability of cyclin D1 and regulated by the phosphorylation of Rb in a CDK/cyclin-dependent way. 33 Our results suggested that PFD decreased the expression of CDK2, CDK6, cyclin D, and cyclin E via the p38 MAPK pathway in HTFs.
Cyclin-dependent kinase inhibitors such as CDKNA1 (p21cip1) and CDKNB1 (P27kip1) can decrease the activation of CDKs/cyclins complexes and thereby induce cell cycle arrest. 34 However, PFD did not have a significant effect on the expression of p21 or p27 (results not shown). The regulation of CDKs and cyclins by PFD may occur through p21- or p27-independent pathways. The specific mechanism needs further investigation.
In this study, we isolated fibroblasts from strabismus surgery other than glaucoma patients based on the following considerations. First, the conjunctive incision in strabismus surgery is located at anterior ocular segment (before equator of eyeball), which is similar to glaucoma surgery. Ethical consideration is the second reason, which is that the SOP of trabeculectomy in China does not include cutting part of Tenon's capsule. To remove part of Tenon's capsule may possibly increase the risks of potential complications, especially when antimetabolites (MMC, 5-FU, etc) are applied during trabeculectomy, such as delayed wound healing (bleb leakage), thin-wall bleb, or even bleb infection. Cutting a small tissue of Tenon's capsules in strabismus patients would not lead to complications the same as in glaucoma surgeries.
Growth factors (GF), such as EGF, FGF, PDGF, and TGF-β, released during the wound healing process can activate the AKT and ERK/MAPK pathways, which are essential for regulating the progression of the G1 phase and fibroblast proliferation. 35 Most studies have revealed that fibroblasts can be activated by TGF-β, which can promote myofibroblast transition. However, regulation of fibroblast proliferation and the cell cycle by TGF-β depends on the cell type or stimulation time. 36 Anti-TGF-β treatment has not prevented the failure of a primary trabeculectomy in a phase III study. This suggests that an antagonist for a single cytokine is not sufficient. 37
Serum obviously activated the proliferation of fibroblasts shown in the results of WST-1 cell proliferation assay and cell cycle analysis. Other authors also reported the similar results about serum-stimulated proliferation of HTFs. 38 Same to our previous study, 21 we selected serum-activated HTFs as a proliferation model to investigate the cell cycle regulation mechanism of PFD and demonstrate a novel underlying profile of the agent to arrest the G1 phase.
We adopted serum-activated fibroblasts rather than TGF-β activated ones, because this study focused on the mechanism of antiproliferative and cell cycle arrest of pirfenidone rather than the single signal pathway of TGF-β. However, it is better to investigate the effect of PFD on the proliferative conditions of HTFs stimulated by serum and TGF-β respectively, which would preferably clarify the mechanism of the antiproliferative and cell cycle arrest effect of this antifibrotic agent. We will investigate and compare the effect of antiproliferative and cell cycle arrest and mechanism in TGF-β and serum-activated fibroblasts in future study.
Conclusions
In summary, PFD inhibited HTFs proliferation and arrested the cell cycle by downregulating CDK6/cyclin D and CDK2/cyclin E involving the inhibition of the AKT/GSK3β, ERK/MAPK, and JNK/MAPK signaling pathways and activation of the p38 MAPK signaling pathway. PFD may be a promising antifibrotic agent in glaucoma filtration surgery.
Footnotes
Acknowledgments
This study was funded by the Natural Science Foundation of China (Grant No. 81300764), the Research Fund for the Doctoral Program of Higher Education of China (Grant No. 20120171130013), and the Pearl River Nova Program of Guangzhou (Grant No. 2016–55).
Author Disclosure Statement
No competing financial interests exist.