
Abstract
Purpose:
To compare the efficacy and safety of HU00701 (0.01% cyclosporin A + 3% trehalose), HU007 (0.02% cyclosporin A + 3% trehalose) (all w/v), and placebo in patients with moderate to severe dry eye disease (
Methods:
This was a multicenter, randomized, double-masked, parallel, placebo-controlled phase II study. In total, 114 patients were randomly assigned to the HU00701, HU007, placebo, or reference group. There was a 2-week run-in period before the 12-week intervention. Efficacy and safety were evaluated every 4 weeks.
Results:
The primary endpoint, change in corneal staining score from baseline to week 12, did not differ significantly among the control, HU00701, and HU007 groups in the full analysis. Of the secondary endpoints, only the tear film breakup time differed significantly at week 12 between the placebo and HU00701 groups. Twenty adverse events were reported by 15 patients, but the rate did not differ significantly among the 4 groups. The laboratory test, vital signs, and physical examination data showed no significant changes during the study.
Conclusions:
HU00701 and HU007 are safe, and HU007 effectively reduces the corneal staining score in patients with moderate-to-severe
Introduction
Dry eye disease (DED) affects the ocular surface and is characterized by loss of tear film homeostasis. The prevalence of DED ranged from 5% to 50% among studies evaluating DE symptoms. 1 When DED is diagnosed based principally on signs, the prevalence rate can reach 75% in certain populations, 2 and it increases significantly with age. However, relatively high prevalence rates have also been reported in younger subjects with risk factors, including intense use of digital devices. As a common condition, DE imposes a financial burden on health care systems and reduces work productivity. 3 DE can be caused by tear film instability and hyperosmolarity, ocular surface inflammation and damage, and neurosensory abnormalities. 4 Medical options include tear replacement, tear stimulators, treatment of lid abnormalities, and anti-inflammatory therapy. 5 Corticosteroids are widely used to treat inflammatory diseases, including DED. However, complications include ocular hypertension, cataracts, and ocular infections.5,6 Cyclosporin A is an anti-inflammatory agent that has no such complications and also exhibits immunomodulatory properties.7,8 The US Food and Drug Administration approved topical cyclosporin A for the treatment of moderate-to-severe DED in 2003. Cyclosporin A decreases the levels of inflammatory mediators, suppresses T cells, and stimulates the accessory lacrimal gland, thus increasing tear production.9,10 However, a major drawback is the burning sensation caused by the eyedrops, which reduces treatment adherence. Moreover, coagulated (cream) products containing non-uniform particles of 0.05% (w/v) cyclosporin A cause blurred vision, irritation, and pain, and they are inconvenient because they must be shaken well before use. Reducing the discomfort and inconvenience would improve treatment adherence.11,12
Thus, we formulated nanoemulsions containing low concentrations of cyclosporin A with 3% (w/v) trehalose to minimize discomfort and inconvenience, increase tolerance, and maximize keratoconjunctival cell protection. Trehalose is a non-reducing disaccharide of glucose that protects against dehydration and oxidative stress, and it stabilizes proteins under stress.13–15 In a preclinical animal study, a mixture of 0.02% cyclosporin A and 3% trehalose increased tear secretion, maintained the goblet cell count, and suppressed corneal damage better than 0.02% cyclosporin A with 1% trehalose or 0.05% cyclosporin A alone (data not shown). In a preclinical study, 0.02% cyclosporin A + 3% trehalose significantly suppressed inflammation and lacrimal gland damage compared with a control group (data not shown). In a preclinical toxicity test using rats, a single subcutaneous injection into the back of the neck was not toxic, whereas a single application of HU007 did not irritate rabbit eyes (Supplementary Tables S1, S2, S3). Therefore, we performed a phase II clinical study to compare the efficacy and safety of HU00701 (0.01% cyclosporin A + 3% trehalose), HU007 (0.02% cyclosporin A + 3% trehalose), and placebo in patients with moderate-to-severe DED.
Methods
Study design
This prospective, randomized, double-masked phase II clinical trial was approved by the Institutional Review Boards of Seoul St. Mary's Hospital, Chonnam National University Hospital, Samsung Medical Center, Daejeon St. Mary's Hospital, and Seoul National University Bundang Hospital. All procedures complied with the tenets of the Declaration of Helsinki, the approved protocol, the International Conference on Harmonization Good Clinical Practice guideline, and all applicable local regulations and laws.
Patients
Male and female patients at least 19 years of age with clinically diagnosed DED were enrolled. At both the screening and baseline visits, both eyes were required to have a corneal staining score (CSS) ≥2 (using the Oxford grading system), 16 a Schirmer score (without anesthesia) ≤10 mm/5 min (if the score was 0 mm/5 min, the score on nasal stimulation was required to be >3 mm/5 min), and a tear film breakup time ≤10 s. Patients were instructed to discontinue all DED medications at the time of their screening visit.
Patients were excluded if they had used a cyclosporin A or steroid ophthalmic solution within 30 days before screening, or if they were currently using (or had recently used) any steroid or non-steroid anti-inflammatory eyedrops or autologous serum eyedrops, had undergone refractive surgery within 12 months before screening, had any ocular disorder or condition that might complicate interpretation of the study results, or were hypersensitive to cyclosporin A or trehalose.
Study protocol
The study included a 2-week run-in period and a 12-week treatment phase. Patients were asked to visit 5 times, including the screening visit (week −3 to −2), the baseline visit (day 0), and 3 more visits at 4-week intervals (week 4, 8, and 12) for assessment of treatment efficacy and safety. All patients gave written informed consent on the day of screening. The participants were instructed to discontinue any DED medications, and to use artificial tears (Hi-Eye Fresh Eyedrops; Huons, Seoul, Korea) whenever needed, during the run-in phase (day −14 to 0). All patients who met the inclusion and exclusion criteria at baseline (day 0) were randomly allocated to the 4 treatment groups in a 2:2:2:1 ratio, that is, the placebo, HU00701 (0.01% cyclosporin A + 3% trehalose), HU007 (0.02% cyclosporin A + 3% trehalose), and reference groups [Restasis (0.05% Cyclosporin A); Allergan, Irvine, CA]. Artificial tear eyedrops could be used up to 3 times a day. Efficacy was assessed at every visit; the “worse eye” (identified during the baseline visit) was used in the efficacy analysis.
Efficacy assessments
The primary efficacy endpoint was the change in CSS between baseline and week 12. Secondary endpoints included the change in CSS between baseline and week 4 and 8, the conjunctival staining scores at week 4, 8, and 12, the total tear volume measured by strip meniscometry (SM), the tear breakup time (TBUT), and the Standard Patient Evaluation of Eye Dryness (SPEED) questionnaire score. The time to 100% clearance and need for rescue medication were assessed throughout treatment.
Corneal and conjunctival staining scores
Corneal and conjunctival damage were estimated at each visit using the Oxford CSS and conjunctival staining score. 16 Sodium fluorescein and lissamine green were used to stain the cornea and conjunctiva, respectively. The Oxford CSS grading system quantifies epithelial surface damage in patients with DED by using a chart with panels labeled A–E (in order of increasing severity). For grading, the colors of the panels are compared with those of the interpalpebral conjunctiva and cornea of the patient.
Strip meniscometry
The SM noninvasively assesses tear meniscus volume. We applied the strip to the lateral lower lid without touching the upper eyelid or ocular surface, and without causing pain. 17 The length (in millimeters) of the stained tear column in the central membrane ditch was the SM value. The SM was performed at all visits except the screening visit.
Tear breakup time
TBUT was measured by instilling 5 μL 2% (w/v) fluorescein solution into the eyes. Patients were asked not to blink under cobalt blue illumination. The TBUT was the time (in seconds) between a normal blink and the appearance of the first dry spot in the tear film. The cut-off for positivity was up to 10 s of subjective observation. 18 Measurements were obtained at each visit, and the mean of 3 measurements was recorded.
SPEED score
The SPEED questionnaire of Korb and Blackie was used to assess the frequency and severity of dry eye symptoms.19,20 The total score ranges from 0 to 28 (8 domains: dryness, grittiness, scratchiness, irritation, burning, watering, soreness, and eye fatigue). The questionnaire also assessed whether the symptoms were non-problematic, tolerable, uncomfortable, bothersome, or intolerable. 21
Time to 100% clearance based on the CSS
The time to 100% clearance was determined. Patients who did not exhibit 100% clearance at the last visit, or who were lost to follow-up, were censored on the last possible day.
Statistical methods
Primary efficacy analysis of the worse eye was performed on the full analysis set (FAS). The FAS included all participants who received at least 1 dose of an investigational product (IP) and underwent at least 1 post-baseline safety assessment. The safety analysis set included all participants who received at least 1 dose of IP.
The CSS, conjunctival staining score, tear quantity, TBUT, and SPEED score were analyzed by the analysis of covariance model (ANCOVA) adjusted for baseline values, to determine inter- and intra-group differences. The adjusted means (least-square means) and P-values are presented. The log-rank and Wilcoxon rank sum tests were used to test for significant between-group differences in the time to 100% CSS clearance and the number of rescue medications used, respectively. All statistical analyses included only the HU00701, HU007, and placebo groups; the reference group was excluded. All adverse events (AEs) were standardized by system organ class and the preferred term by using the Medical Dictionary for Regulatory Activities (MedDRA; ver. 18.0). The significance of between-group differences in AE incidence rates was tested by using the chi-squared or Fisher's exact test. Vital signs and laboratory test data were compared by using the paired t-test or Wilcoxon signed-rank test. ANOVA or the Kruskal–Wallis test was used to compare laboratory and physical examination data before and after treatment. All analyses were performed by using SAS software (ver. 9.4; SAS Institute, Cary, NC), and the significance level was set to 5% (2-sided).
Results
Of the 142 patients recruited from the 5 hospitals, 114 were enrolled and randomly assigned to the placebo group (n = 34), HU00701 group (n = 32), HU007 group (n = 31), or reference group (n = 17; Fig. 1). In total, 97% of patients in the placebo group, 97% in the HU00701 group, 100% in the HU007 group, and 88% in the reference group completed the study; their demographic characteristics are summarized in Table 1 (intergroup differences are not shown). Deviations from the study protocol were assessed before unmasking. Major deviations included violation of the inclusion and exclusion criteria, the use of prohibited concomitant medication, and noncompliance. In total, 10 deviations were classified as major: 5 were related to test drugs, 3 to the visit window, 1 to noncompliance, and 1 to violation of the inclusion criteria.
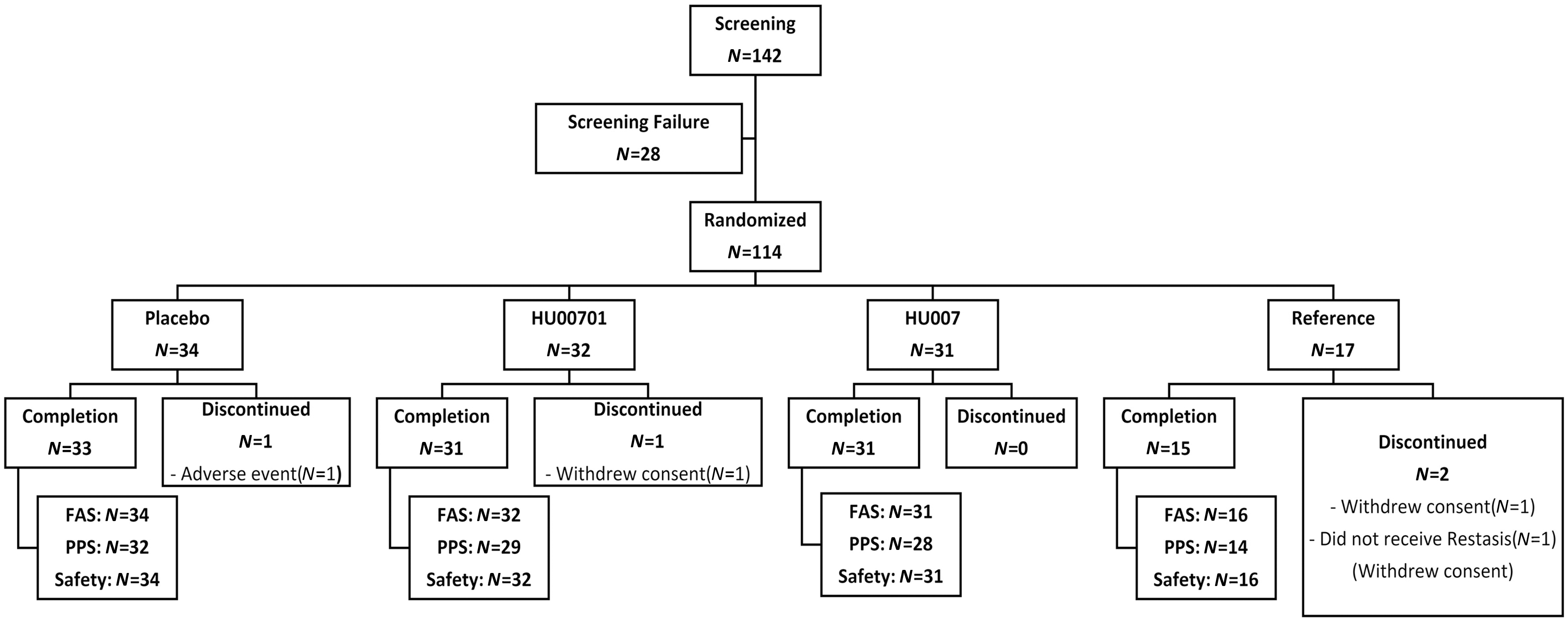
Flowchart showing the patient group allocations.
Baseline Patient Demographic Characteristics
Kruskal–Wallis test.
Chi-squared test.
NC, not calculated; SD, standard deviation; TBUT, tear film break-up time.
Efficacy
Primary endpoint
The primary endpoint was the mean change in the CSS between baseline and week 12. Efficacy was determined in the FAS groups. The reference group was not included in the analyses. In the FAS population, CSSs at baseline were similar among all groups, with means ± standard deviations of 2.21 ± 0.48 in the placebo group, 2.16 ± 0.37 in the HU00701 group, and 2.16 ± 0.37 in the HU007 group. The changes in CSSs after 12 weeks of treatment were −1.31, −1.48, and −1.68 in the placebo, HU00701, and HU007 groups, respectively (all P < 0.0001). The differences from the placebo group were −0.16 (P = 0.39) in the HU00701 group and −0.37 (P = 0.06) in the HU007 group (Fig. 2).

Mean CSSs of patients with
Secondary endpoints
CSS at week 4 and 8
The FAS analysis showed changes in CSSs after 4 and 8 weeks of treatment of −0.75 and −1.08 in the placebo group, respectively (both P < 0.0001); the respective values in the HU00701 group were −0.85 and −1.35 (both P < 0.0001), and those in the HU007 group were −0.91 and −1.33 (both P < 0.0001) (Fig. 2). The differences from the placebo group were −0.10 (P = 0.56) at week 4 and −0.27 (P = 0.12) at week 8 in the HU00701 group; and −0.16 (P = 0.36) at week 4 and −0.25 (P = 0.16) at week 8 in the HU007 group.
Conjunctival staining score at weeks 4, 8, and 12
The FAS analysis showed that the changes in conjunctival staining scores after 4, 8, and 12 weeks of treatment were −0.86, −0.89, and −0.94 in the placebo group (all P < 0.0001); −0.43, −1.20, and −1.16 in the HU00701 group (all P < 0.05); and −0.65, −1.05, and −1.32 in the HU007 group, respectively (all P < 0.001)(Fig. 3). The differences from the placebo group were 0.43 (P = 0.11) at week 4, −0.31 (P = 0.19) at week 8, and −0.22 (P = 0.44) at week 12 in the HU00701 group; and 0.21 (P = 0.42) at week 4, −0.16 (P = 0.49) at week 8, and −0.37 (P = 0.19) at week 12 in the HU007 group. Both the HU00701 and HU007 groups exhibited somewhat larger decreases at week 8 and 12 compared with the placebo group, but the differences were not significant.
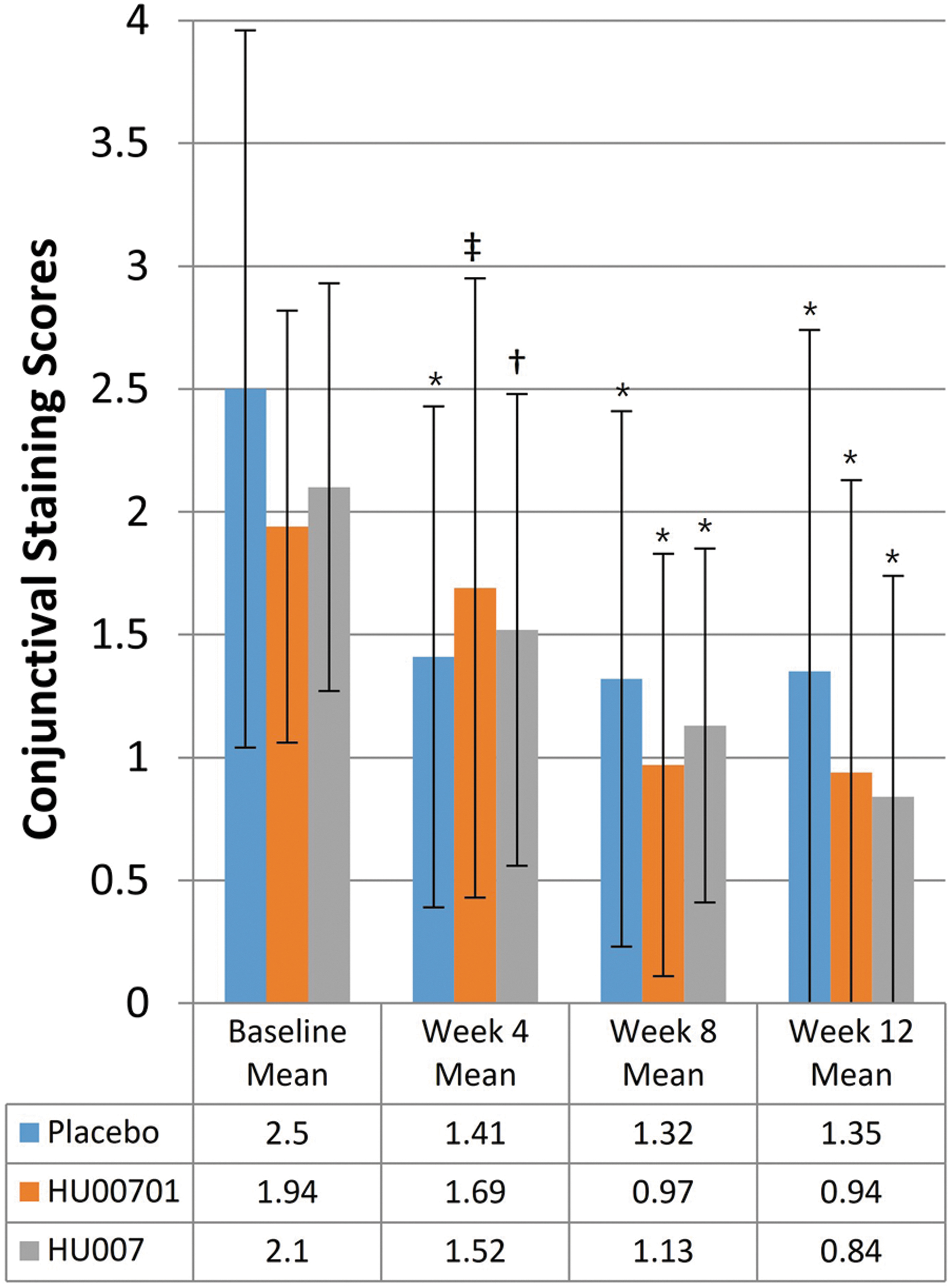
Mean conjunctival staining scores of patients with DED receiving placebo, HU00701, or HU007 treatment for 12 weeks. Conjunctival staining scores were assessed by using the Oxford grading system. *P < 0.0001,
SM at weeks 4, 8, and 12
The FAS analysis showed that the changes in tear volume after 4, 8, and 12 weeks of treatment were −0.15, −0.10, and −0.04 mm/5 s in the placebo group (P = 0.60, 0.81, and 0.91, respectively); −0.06, 0.09, and −0.23 mm/5 s in the HU00701 group (P = 0.84, 0.84, and 0.47, respectively); and −0.39, 0.38, and 0.18 mm/5 s in the HU007 group, respectively (P = 0.19, 0.39, and 0.58, respectively). The differences from the placebo group were 0.09 mm/5 s (P = 0.82) at week 4, 0.19 mm/5 s (P = 0.76) at week 8, and −0.20 mm/5 s (P = 0.66) at week 12 in the HU00701 group; and −0.24 mm/5 s (P = 0.56) at week 4, 0.48 mm/5 s (P = 0.43) at week 8, and 0.22 mm/5 s (P = 0.63) at week 12 in the HU007 group.
Tear film breakup times at weeks 4, 8, and 12
The FAS analysis showed that the respective changes in TBUTs after 4, 8, and 12 weeks of treatment were 0.57, 0.77, and 0.48 s in the placebo group (P = 0.05, 0.07, and 0.13); 0.86, 1.51, and 1.58 s in the HU00701 group (all P < 0.01); and 0.68, 1.63, and 1.04 s in the HU007 group (all P < 0.05). The differences from the placebo group were 0.29 s (P = 0.48) at week 4, 0.74 s (P = 0.22) at week 8, and 1.10 s (P = 0.02) at week 12 in the HU00701 group; and 0.11 s (P = 0.79) at week 4, 0.86 s (P = 0.16) at week 8, and 0.55 s (P = 0.23) at week 12 in the HU007 group. The HU00701 group exhibited a significant increase at week 12 compared with the placebo group.
SPEED questionnaire scores at weeks 4, 8, and 12
The FAS analysis showed that the respective changes in SPEED scores after 4, 8, and 12 weeks of treatment were −4.48, 4.92, and −5.04 in the placebo group (all P < 0.0001); −4.72, −5.55, and −6.15 in the HU00701 group (all P < 0.0001); and −4.09, −4.78, and −5.20 in the HU007 group (all P < 0.0001) (Fig. 4). The differences from the placebo group were −0.24 (P = 0.78) at week 4, −0.63 (P = 0.52) at week 8, and −1.11 (P = 0.23) at week 12 in the HU00701 group; and 0.39 (P = 0.66) at week 4, 0.14 (P = 0.87) at week 8, and −0.16 (P = 0.86) at week 12 in the HU007 group.
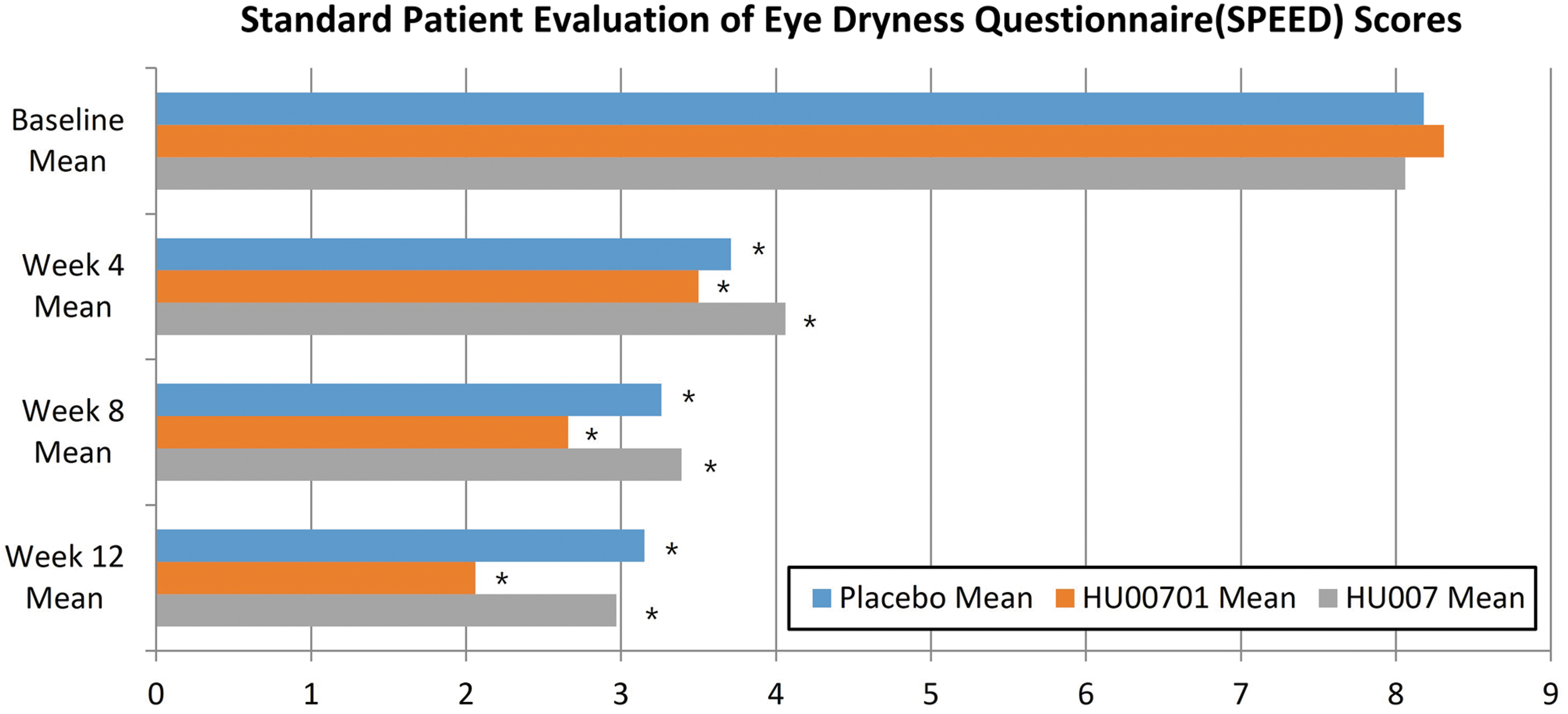
Mean SPEED scores of patients with DED receiving placebo, HU00701, or HU007 treatment for 12 weeks. *P < 0.0001 compared with baseline. SPEED, Standard Patient Evaluation of Eye Dryness.
Time to reach 100% corneal clearance
The FAS analysis showed that the median times to 100% corneal clearance were 86.0 days in 17 patients in the placebo group (50.0%), 85.0 days in 16 patients in the HU00701 group (50.0%), and 85.0 days in 23 patients in the HU007 group (74.19%). Although more patients achieved 100% clearance in the HU007 than in the placebo group, the difference was not significant (HU00701 group P = 0.92; HU007 group P = 0.14).
Numbers of rescue medications
The FAS analysis showed that the median numbers of rescue medications used during treatment were 26.5, 66.0, and 10.0 in the placebo, HU00701, and HU007 groups, respectively. The median numbers of rescue medications used per week were 2.17, 5.28, and 0.71, respectively, and they did not differ significantly among the groups (HU00701 group P = 0.14; HU007 group P = 0.82).
Safety
In total, 133 patients were included in the safety set. In total, 20 AEs were reported by 15 patients: 3 by patients in the placebo group (n = 3, 8.82%), 5 by patients in the HU00701 group (n = 4, 12.50%), 8 by patients in the HU007 group (n = 4, 12.90%), and 4 by patients in the reference group (n = 4, 25.00%) (Table 2). The AE incidence did not differ significantly among the groups. Nine AEs were mild, 10 were moderate, and 1 was severe. One AE (eye pain) in the placebo group was mild and not related to the study. The severe AE (a contusion) occurred in an HU007 group patient, and it was not related to the study treatment. No clinically significant change in, or abnormality of, any vital sign or laboratory test was evident after IP administration.
Incidence of Treatment-Emergent Adverse Events by System Organ Class on the Safety Set
Treatment-emergent adverse events are displayed as number of patients (percentage) [number of events].
Discussion
This randomized, placebo-controlled phase II clinical trial assessed the efficacy and safety of 0.01% or 0.02% cyclosporin A with 3% trehalose in patients with moderate-to-severe DED. On FAS analysis, the changes in the primary endpoint (change in CSS at week 12) were somewhat larger in the HU00701 and HU007 groups than in the placebo group, but the differences were not significant. No drug-related AEs were observed in the HU00701 or HU007 group. No participant exhibited a clinically significant change or abnormality in vital signs or laboratory data.
The HU00701 and HU007 groups exhibited significant improvements in the CSS, conjunctival staining score, tear breakup time, and SPEED score compared with baseline. The staining score improvements were evident by week 4 in both groups and persisted to week 12. HU00701 and HU007 were at least somewhat efficacious based on the results cited earlier. The HU007 group achieved 100% clearance more rapidly than did the placebo group, and the weekly number of rescue medications was lower than in the other groups, although not significantly.
Since the introduction of 0.05% cyclosporin A to treat DED, new drugs varying in terms of the cyclosporin A concentration or formulation have undergone clinical trials.22–24 Goldberg et al. tested a clear, aqueous nanomicellar formulation of 0.09% cyclosporin A in DED patients. Although DED improved, 24.2% of the patients receiving the formulation complained of instillation site pain compared with only 4.3% of the vehicle group. Thus, high-dose cyclosporin A may trigger AEs, which reduce treatment adherence. In a large study of patients with glaucoma,25–27 Kim et al. reported that approximately one-third of 1,046 patients were non-adherent. 25 One retrospective study on DED patients treated with a 0.05% cyclosporin A ophthalmic emulsion found that the adherence rate was only 5.9%, with a median time to discontinuation of 3 months. 28 A meta-analysis of 12 randomized controlled trials using topical 0.05% cyclosporin A reported that the treatment-related AE rate was higher than in controls 29 ; the most common AEs were ocular burning and stinging. 30 Thus, the dose of cyclosporin A in our eye drops was lower, to decrease the rate of AEs and increase adherence.
Trehalose is a non-reducing disaccharide of glucose that is synthesized by many organisms, including bacteria, yeast, fungi, insects, invertebrates, and plants. 31 Its unique properties include protection against dehydration and oxidative stress, and stabilization of proteins under stress.13–15 Numerous prospective studies have found that trehalose protects the ocular surface in DED patients. When used to treat moderate-to-severe DED, trehalose solutions were better than commercial eyedrops containing hyaluronan and hydroxyethylcellulose. 32 Wozniak et al. tested the effect of 3 lubricant eye gels on tear film thickness (TFT). 33 Eyedrops containing 3% trehalose and 0.15% hyaluronic acid (HA) significantly improved TFT compared with eyedrops with 0.2% HA or 0.4% polyethylene glycol and 0.3% propylene glycol. Moreover, eyedrops with 3% trehalose and 0.15% HA remained on the tear film longer compared with other eyedrops. In another randomized controlled study comparing efficacy and safety between HA-trehalose eyedrops and HA eyedrops, Chiambaretta et al. concluded that the former were more effective, safer, and better accepted by patients. 34 In a randomized, single-center open-label crossover study on patients with moderate-to-severe DED receiving a combination HA-trehalose treatment or Systane, the former treatment was at least as effective as the latter overall, and more effective in certain respects. 35 Thus, trehalose may be beneficial when combined with HA.
In this study, we combined trehalose with cyclosporin A and evaluated its efficacy and safety. Although our results seem promising, there were certain limitations. First, the minimum therapeutic dose of cyclosporin A for DED remains unclear, although the HU007 group exhibited somewhat larger improvement in the CSS. In a phase II study of 0.05, 0.1, 0.2, and 0.4% cyclosporin A, Stevenson et al. found no clear dose-response relationship, and they recommended both the 0.05 and 0.1% formulations for future clinical studies. 36
Second, any synergistic effects of trehalose and cyclosporin A remain unclear, although in previous studies trehalose augmented the effects of HA on DED.34,35 As trehalose interacts with other compounds, it may enhance the effectiveness of cyclosporin A. Further pharmacokinetic and pharmacodynamic data from human trials are required.
Third, as this was a phase II study, there was no active competitor; Restasis (0.05% cyclosporin A) served as the reference. We aim at addressing this in our planned phase III study. However, a concern here is that a double-masked design could not be used, because the HU00701 and HU007 doses differ from that of Restasis, which is the only comparator approved for use in clinical trials of cyclosporin A eyedrops in South Korea. Therefore, this problem is unavoidable, as reported in previous studies.30,37
Conclusions
This phase II clinical study showed that HU00701 and HU007 eyedrops were safe, and it also suggested that HU007 was clinically efficacious for treating DED. A larger cohort will be required for our planned phase III clinical trial.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.