
Abstract
Purpose:
The fluid pump and barrier functions of the corneal endothelium maintain stromal deturgescence required for corneal transparency. The effect of oxidative stress, a hallmark of Fuchs endothelial corneal dystrophy (FECD), on the endothelial barrier function has been investigated.
Methods:
The endothelium of porcine corneas ex vivo was exposed to (1) membrane permeable oxidants (H2O2, 100 μM, 1 h; tert-butyl-hydroperoxide, 100 μM, 1 h), or (2) ultraviolet A (UVA) with photosensitizers for 15 min, riboflavin (50 μM) or tryptophan (Trp) (100 μM). The effects on the apical junction complex were analyzed by (1) immunostaining the perijunctional actomyosin ring (PAMR) and ZO-1 and (2) assessment of paracellular flux of fluorescein isothiocyanate (FITC)–avidin across cultured endothelial cells grown on biotinylated-gelatin film. The extent of oxidative stress was quantified by changes in intracellular reactive oxygen species (ROS) and mitochondrial membrane potential (MMP) in addition to lipid peroxidation and release of lactate dehydrogenase (LDH).
Results:
Both methods of oxidative stress led to the disruption of PAMR and ZO-1 concurrent with changes in ROS levels, depolarization of MMP, increased lipid peroxidation, elevated LDH release, and increased permeability of FITC-avidin. The effects of direct oxidants were opposed by SB-203580 [p38 mitogen-activating protein (MAP) kinase inhibitor; 10 μM]. The damage by UVA+photosensitizers was blocked by extracellular catalase (10,000 U/mL).
Conclusions:
(1) Acute oxidative stress breaks down the barrier function through destruction of PAMR in a p38 MAP kinase-dependent manner. (2) UVA+photosensitizers elicit the breakdown of PAMR via type I reactions, involving H2O2 release. (3) Blocking the oxidative stress prevents loss of barrier function, which could be helpful in the therapeutics of FECD.
Introduction
Fuchs endothelial corneal dystrophy (FECD), a progressive and late-onset genetic disorder, affects ∼4% of individuals aged >40 years in the United States. 1 At present, the standard of care is corneal transplantation because no pharmacological approaches exist to treat the disorder. This is because the pathophysiology of the disease remains incompletely understood.1–3 However, the main hallmarks of the disorder include accelerated apoptotic cell death (leading to reduced endothelial cell density or ECD), accumulation of extracellular matrix, oxidative stress, and endoplasmic reticulum (ER) stress.1,3–5 Oxidative stress is also implicated in the pathogenesis of other endothelial disorders, including Pseudophakic Bullous Keratopathy and Congenital Hereditary Endothelial Dystrophy,6–8 in addition to FECD. In this study, we have focused on the effects of acute, experimental oxidative stress on the barrier function of the corneal endothelium.
The barrier function of the corneal endothelium is the resistance to a persistent fluid leak from the anterior chamber to the stroma.9,10 Thus, the restraint to the fluid influx supports the relative state of dehydration (stromal deturgescence) required for corneal transparency. The fluid pump activity balances the barrier function by returning the residual fluid leak from the stroma into the anterior chamber. 9 The barrier function is conferred by the tight junctions of the endothelium. 10 As in many epithelial and vascular endothelial monolayers, the integrity of tight junctions is influenced by a tuft of the actin cytoskeleton at the apical junctional complex (AJC), referred to as the perijunctional actomyosin ring (PAMR) 11 (also reviewed by Srinivas9,10). In fact, PAMR regulates the integrity of both adherens and tight junctions and vice versa in the endothelium.9,12
The integrity of PAMR is regulated by various cell signaling pathways, including the RhoA-Rho kinase axis and p38 mitogen-activating protein (MAP) kinase.9,10,12–14 Thus, activating Rho kinase by exposure to thrombin breaks down the tight junctions by increasing the contractility of PAMR.9,10,13 The contraction generates a centripetal force that pulls the tight junctions apart, causing a breakdown of the barrier integrity.10,12 In addition, activation of p38 MAP kinase, downstream of tumor necrosis factor α (cytokine stress) 14 and hypothermia (cold stress) 15 also adversely affects the integrity of PAMR and hence breaks down the tight junctions. Overall, stress signaling pathways affect the barrier function through their influence on PAMR. In particular, oxidative stress is known to affect the actin cytoskeleton through many mechanisms, including microtubule disassembly.16–23 Therefore, it is imperative to examine the effects of oxidative stress on the barrier function.
We recently reported the effects of acute oxidative stress on the barrier function in cultured porcine corneal endothelial cells. 24 The barrier function was measured by TER (trans-endothelial resistance), whereas the corresponding destruction of the AJC was imaged by immunocytochemistry. Specifically, we reported that experimental oxidative stress induces microtubule disassembly, leading to a breakdown of PAMR and barrier function. We also demonstrated that the impact on the barrier function was attenuated by SB-203580, a p38 MAP kinase inhibitor, indicating a role for the stress kinase. 24 In this study, we have extended our investigation to the endothelium associated with ex vivo corneas. As in the previous study, we have found that acute oxidative stress disrupts PAMR and ZO-1, leading to a breakdown of the barrier integrity. Concurrently, the oxidative stress elevated reactive oxygen species (ROS) levels, depolarized mitochondrial membrane potential (MMP), increased lipid peroxidation, elevated lactate dehydrogenase (LDH) release, and increased local permeability of fluorescein isothiocyanate (FITC)-avidin.
Methods
Chemicals and antibodies
4′,6-diamidino-2-phenylindole (DAPI) (D9542), 0.4% Trypan blue (T8154), attachment factor (123–100), antibiotic-antimycotic (A5955), SB-203580 (S8307), antifade reagent (P36930),
Oxidative stress induced in ex vivo corneas
Fresh porcine eyes were obtained from a local slaughterhouse and placed in a povidone–iodine solution [1% in phosphate-buffered saline (PBS)] for 5 min. The eyeballs were then rinsed with 1 × PBS and corneal buttons were isolated. The corneas were then held everted on Petri dishes.
For experimental oxidative stress, we used 2 methods. In the first method, we exposed the endothelial surface to H2O2 (100 μM; 1 h) or ter-butyl-hydroxyperoxide (tBHP) (100 μM; 1 h) with and without pre- and co-treatment with SB-203580 (10 μM; 1 h). In the second approach, we induced acute oxidative stress by exposing the endothelial surface bathed in a medium containing riboflavin (Rf; 50 μM) or Trp (100 μM) to ultraviolet A (UVA) (365 nm) for 15 min. In some experiments, the bathing medium contained catalase (10,000 U/mL). In addition, as a positive control for disruption of the barrier function, the endothelial surface was exposed to Ca2+-free medium [with 2 mM ethylene glycol-bis-N,N,N′,N′-teraacetic acid (EGTA)] for 5 min.
Immunocytochemistry
After the termination of oxidative stress, the endothelial surface was quickly rinsed with PBS, and the whole tissue was fixed with 4% paraformaldehyde for 20 min. Subsequently, the endothelium was permeabilized using 0.5% Triton X-100 (in PBS) for 2 min at room temperature. Next, PAMR was stained with phalloidin conjugated to Texas red (1:400) for 1 h at room temperature. Finally, nuclei were stained with DAPI.
After fixation and permeabilization, the endothelial layer was immunostained in separate corneas for ZO-1. Specifically, the tissue was blocked with Tris-buffered saline containing 2% goat serum for 1 h and then incubated with ZO-1 primary antibody (1:20) at 4°C. After 18 h of incubation, the primary antibody was washed off with PBS (3 × ). Next, the endothelial surface was exposed to goat anti-mouse IgG with Alexa fluor-488 at a concentration of 1:700 (secondary antibody). Next, the endothelium was exposed to DAPI for 10 min to stain nuclei.
After staining for PAMR or ZO-1, the tissue was rinsed with PBS and mounted on a glass slide using an antifade reagent on the endothelial side. Fluorescence images were captured at 63 × objective (oil; 1.4 Numerical Aperture) using a Zeiss LSM 880 confocal microscope.
Cell culture
For permeability, ROS, and MMP assays, we cultured porcine corneal endothelial cells as described previously. 24 Isolated corneal buttons obtained as described previously were placed everted in a Petri dish, and the endothelial layer was bathed in PBS containing 0.4% Trypan blue. Under a microscope, snippets of the endothelium (with Descemet's membrane) were peeled off. Next, the explants were cultured on Petri dishes precoated with the attachment factor using DMEM supplemented with 15% fetal bovine serum and 1% antibiotic–antimycotic solution in a CO2 incubator at 37°C. The endothelial cells migrated from the explants to the culture plates in ∼4 days, leading to 90% confluence in 8–10 days.
Permeability assay using FITC-avidin
We measured the local permeability of endothelial cells using FITC-avidin to assess loss of barrier function based on the protocol established by Dubrovskyi et al. 25 Cultured endothelial cells were seeded (10,000–50,000 cells/coverslips) on coverslips precoated with biotinylated gelatin (250 μL of 0.25 mg/mL) and grown for 48–72 h to reach confluence. The monolayers were then exposed to H2O2 (100 μM; 1 h), UVA (365 nm; 15 min), or Rf+UVA (Rf; 50 μM) while being cultured in DMEM containing 2% serum. In some experiments, pre- and co-treatment with SB-203580 and catalase were carried out.
After the termination of the oxidative stress, FITC-avidin (25 μg/mL) was added directly to the medium for 15 min. Next, the monolayer was washed with PBS 2 × and fixed with 3.7% paraformaldehyde for 10 min, followed by staining for PAMR with phalloidin conjugated to Texas red (1:400; 1 h at room temperature). Subsequently, the nucleus was stained with a final concentration of 1 μg/mL DAPI for 10 min. Finally, stained cells were washed using PBS, mounted on glass slides using an antifade reagent, and visualized using Zeiss LSM 880 confocal microscope with the 63 × objective. The assay was repeated with 3 independent preparations of the endothelial monolayers.
LDH assay
The release of LDH enzyme from the endothelium was assayed using the EZcount™ LDH assay kit. 26 First, the endothelial surface of porcine corneas ex vivo was exposed to oxidative stress by the 2 approaches (1) H2O2 or tBHP and (2) photodynamic approach (ie, UVA, Rf+UVA or Trp+UVA) with and without pre- and co-treatment of SB-203580 and catalase. Triton X-100 was used as a positive control for LDH release. Following the termination of the oxidative stress, the endothelial monolayer was scraped off along with the medium and lysed for 30 min (using lysis buffer provided along with the kit). The resulting lysate was centrifuged at 10,000 rpm for 10–15 min at 4°C. Next, the supernatant was mixed with the LDH assay buffer and incubated for 30 min for color development.
Soon after incubation, absorbance at 450 nm was read using a UV-Vis spectrophotometer (Shimadzu-1800, Kyoto, Japan). The LDH release was calculated using the formula %LDH release = 100 × (A − C)/(B − C), where A is the average absorbance of samples after oxidative stress (treated), B is the average absorbance of untreated samples (control), and C is the average absorbance of background (media alone), respectively. The assay was repeated with 3 corneal buttons.
Lipid peroxidation assay
Malondialdehyde (MDA) production under oxidative stress was detected as a marker of lipid peroxidation. 27 The endothelial surface of porcine corneas ex vivo was exposed to various treatments as mentioned previously, and then the endothelial layer was scraped off from the cornea, lysed using a lysis buffer solution (provided with the kit), and incubated for 30 min at room temperature. The lysate was collected following centrifugation (10,000 rpm for 20 min at 4°C). The supernatant was collected and assayed for released MDA by mixing with the assay reagent. The absorbance was read at 530 nm using a UV-Vis spectrophotometer. The assay was repeated with 3 corneal buttons.
ROS assay
ROS production in response to experimental oxidative stress was detected in cultured cells using dichlorodihydrofluorescein diacetate (DCF-DA) EZAssay™ Reactive oxygen species assay kit. 28 Following the termination of treatments, cells were incubated with DCF-DA (final concentration of 2 μM) at 37°C in a CO2 incubator. After 30 min of incubation, cells were washed with PBS, and cellular fluorescence was read (λex = 488 ± 10 nm; λem = 528 ± 10 nm) using Infinite 200 PRO Tecan multimode fluorescence microplate reader. The assay was repeated with 3 independent preparations of the endothelial monolayers.
MMP measurement
MMP was measured using JC-1 (EZAssay Mitochondrial Membrane Potential Assay kit; Himedia). 28 Cells were cultured to 90% confluence on 96-well plates and exposed to different treatments as mentioned previously. After terminating the oxidative stress by washing with PBS, cells were exposed to JC-1 (2 μM final concentration) and incubated in a CO2 incubator. After 30 min of incubation, cells were rinsed with PBS and ratio of fluorescence of JC-1 was recorded (for red fluorescence; λex = 550; λem = 600 nm and green fluorescence; λex = 485; λem = 535 nm) using Infinite 200 PRO Tecan multimode fluorescence microplate reader. The assay was repeated with 3 independent preparations of the endothelial monolayers.
Statistical analysis
One-way analysis of variance with Tukey's post-test analysis was used for statistical comparisons using the GraphPad Prism™ software (version 5.0; San Diego, CA). Data are expressed as mean ± SE. “n” represents the number of independent experiments.
Results
We have examined the impact of oxidative stress on the barrier integrity of ex vivo corneal endothelium by immunocytochemical imaging of AJC. 24 Specifically, we have imaged PAMR and ZO-1 that colocalize at the AJC. In addition, we have measured the permeability to FITC-avidin (68 kDa). We also verified the onset of oxidative stress by measuring intracellular ROS levels and other markers of oxidative stress, including lipid peroxidation, altered MMP, and LDH release.
Impact of extracellular Ca2+ depletion on barrier integrity
We first demonstrate that the loss of barrier integrity in the endothelium is coincident with the destruction of PAMR as a benchmark to further assess the impact of acute oxidative stress. In this approach, we exposed the endothelial surface to Ca2+-free medium (containing EGTA, 2 mM; 5 min). The consequent depletion of extracellular Ca2+ is expected to break down the AJC via disruption of Ca2+-dependent adherens junctions (AJs).12,29
In the untreated corneas, as shown previously in the cat corneal endothelium, 30 the endothelial PAMR shows a distinct honeycombed organization (Fig. 1A, white arrows). Because ZO-1 and PAMR interact, the former shows colocalization contiguously at the cellular periphery. This marks an intact barrier integrity of the endothelium (Fig. 1B). 9 When the endothelium was exposed to a Ca2+-free medium (with EGTA, 2 mM; 5 min), PAMR undergoes marked contraction as shown by circular bundling of F-actin and retraction of ZO-1 (Fig. 1C, D; shown by yellow arrows). The consequent contraction results in intercellular gaps, evident by discontinuous ZO-1 staining (Fig. 1D), indicating a breakdown of the barrier integrity. In some cells, there was even marked loss of ZO-1 staining, possibly owing to endocytosis of the molecule (Fig. 1D; shown by yellow arrows).
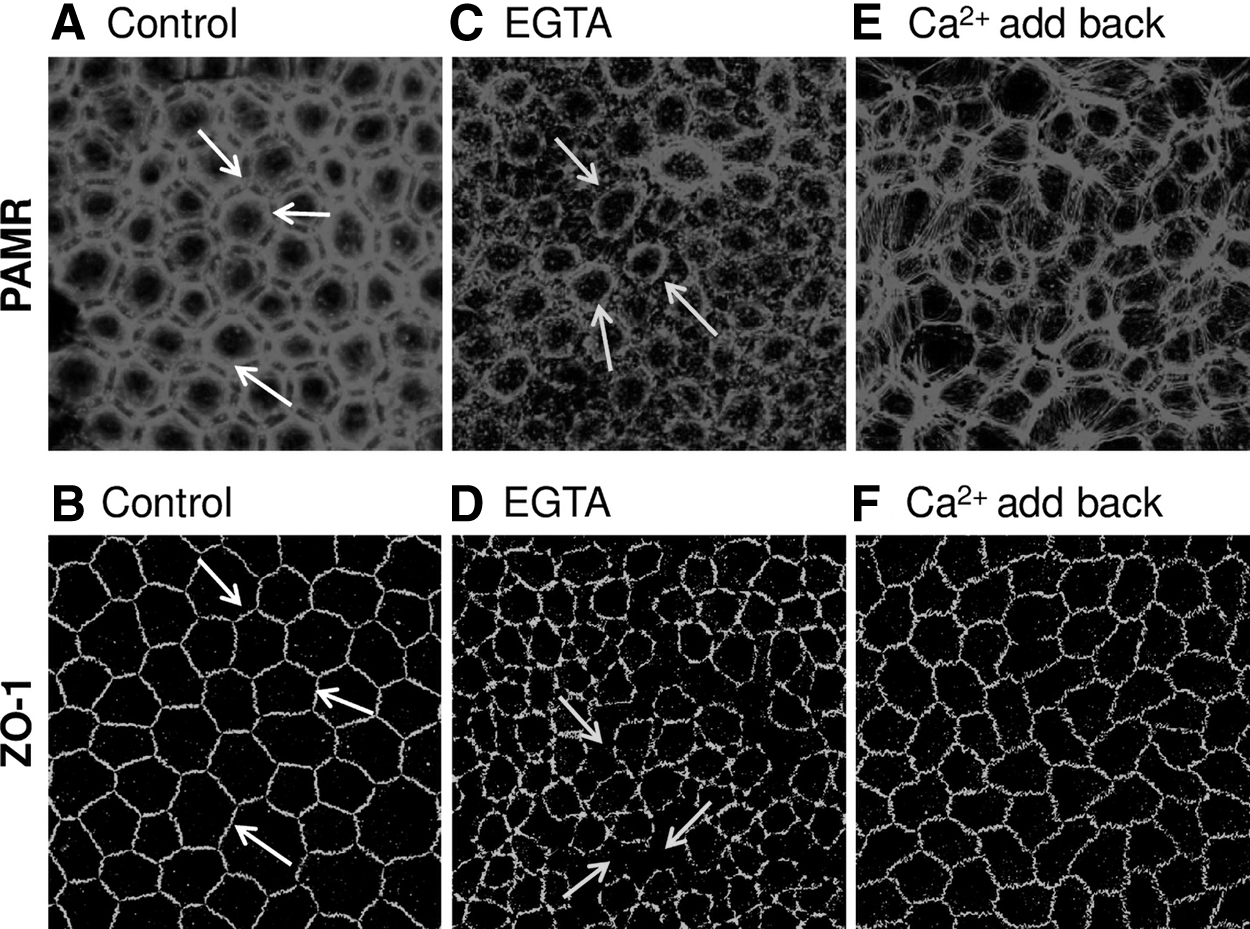
Effect of extracellular Ca2+ depletion on PAMR and ZO-1. The endothelial surface was exposed to a Ca2+-free medium (containing 2 mM ethylene glycol-bis-N,N,N′,N′-teraacetic acid) for 5 min at 37°C with or without returning to the Ca2+-rich medium for 3 h (Ca2+ add-back). Following the treatments, corneas were fixed for immunocytochemistry and stained for endothelial PAMR and ZO-1.
As shown previously, a Ca2+ add-back following Ca2+ depletion maneuver is expected to reanneal AJs and reform the tight junctions. 12 As given in Fig. 1E and F, organization of PAMR and ZO-1 returned partially toward the normal, that is, hexagonal morphology of PAMR and continuous ZO-1 in response to Ca2+ add-back (1.8 mM; 3 h). These experiments show that imaging PAMR at the focal plane of ZO-1 is sufficient to delineate the breakdown of AJCs and thus establish our protocols for investigating the oxidative stress response in ex vivo corneas. In addition, the data in Fig. 1 recapitulate our previous observations with cultured cells 12 and recent study indicating the loss of barrier function of the endothelial cells during corneal cold storage. 15
Impact of acute oxidative stress by extracellular H2O2 and tBHP
We began evaluating oxidative stress by exposing the endothelial surface of ex vivo porcine corneas to membrane permeable oxidants, H2O2 (100 μM) or tBHP (100 μM) at 37°C for 1 h. The endothelial surface was also pre- and co-treated with SB-203580 (10 μM; 1 h), a potent p38 MAP kinase inhibitor. When exposed to H2O2 or tBHP, the organization of PAMR and ZO-1 of the endothelium was destroyed (Fig. 2C, 2D, 2I, and 2J; shown by yellow arrows). The dispersion of ZO-1 at the cell border (pointed by arrows in Fig. 2D, J) indicates a breakdown of barrier integrity. In contrast, the damage to PAMR and ZO-1 was attenuated upon pre- and co-treatment with SB-203580 (Fig. 2G, H, K, L). SB-203580 was not toxic, as shown by a lack of damage to PAMR and ZO-1 (Fig. 2E, F). These results indicate that activation of p38 MAP kinase is involved in H2O2 and tBHP-induced damage to AJC in the endothelium. These results are similar to our previous findings with cultured cells. 24
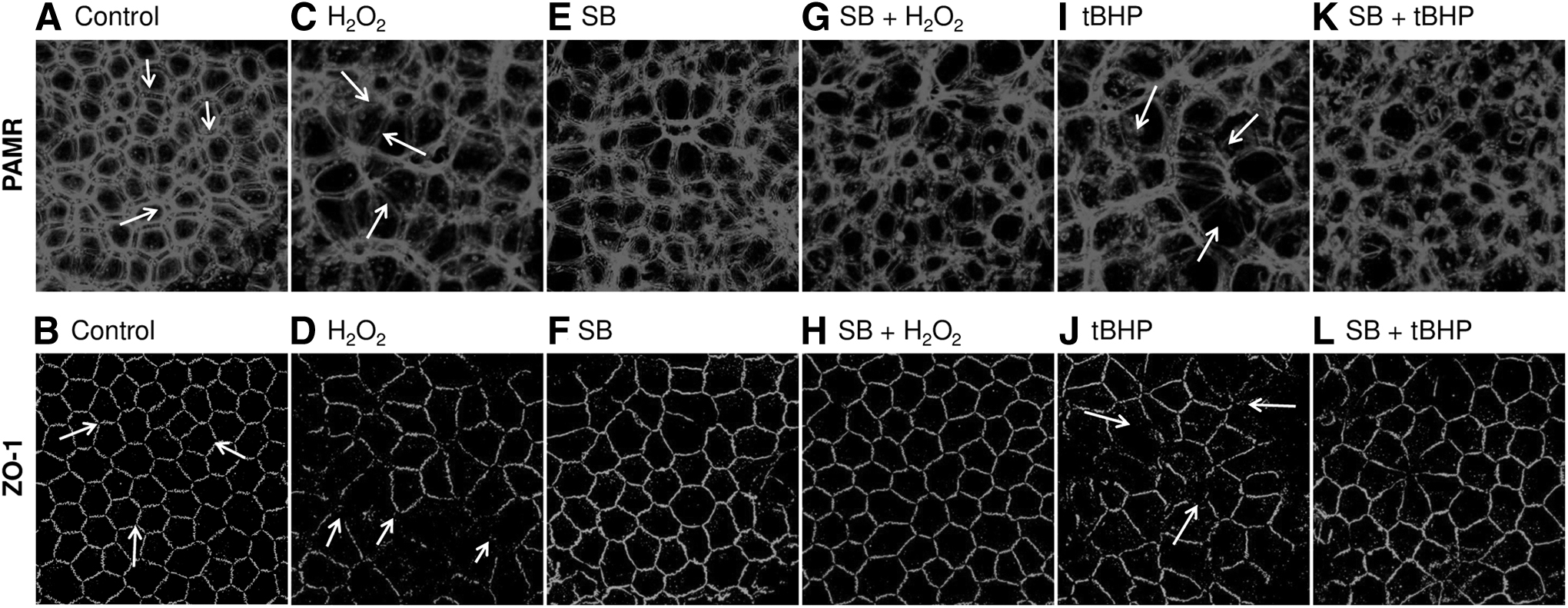
Effect of H2O2 and tBHP on PAMR and ZO-1. The endothelial surface was exposed to H2O2 (100 μM) or tBHP (100 μM) at 37°C for 1 h with and without pre- and co-treatment with SB-203580 (SB; 10 μM, 1 h; a p38 mitogen-activating protein kinase inhibitor). Following the treatments, endothelial PAMR and ZO-1 were immunostained.
Impact of oxidative stress by photodynamic stress
Previously we have established an alternative approach for the induction of acute oxidative stress in the endothelium. 24 In particular, we elicited oxidative stress by induction of type-I photodynamic reactions in situ using Rf as a photosensitizer. 24 Specifically, we exposed cultured endothelial cells bathed in a culture medium containing 2% serum and 50 μM Rf to UVA (365 nm; 15 min). In this study, we have extended the approach to the endothelium associated with intact corneas ex vivo.
Figure 3C and D shows the destruction of PAMR and ZO-1 by UVA alone. However, the destruction is pronounced in response to Rf+UVA (Fig. 3E, F). Because type-I reactions elicit H2O2 release, the effect of Rf+UVA is akin to the direct exposure of H2O2 in Fig. 2C and D. Thus, including catalase in the bathing medium before exposure to Rf+UVA completely blocked the destruction of AJC (Fig. 3G, H). As an alternative to Rf, we also used Trp as the photosensitizer. In particular, Trp elicits type-I photochemical reactions in response to UVA. 31 As shown in Fig. 3I and J, the effect of Rf+UVA is recapitulated by Trp+UVA. Similarly, the inclusion of catalase before Trp+UVA also limited the destruction of AJC (Fig. 3K, L). These results are reminiscent of immunocytochemical and TER findings that we have reported previously with cultured cells. 24
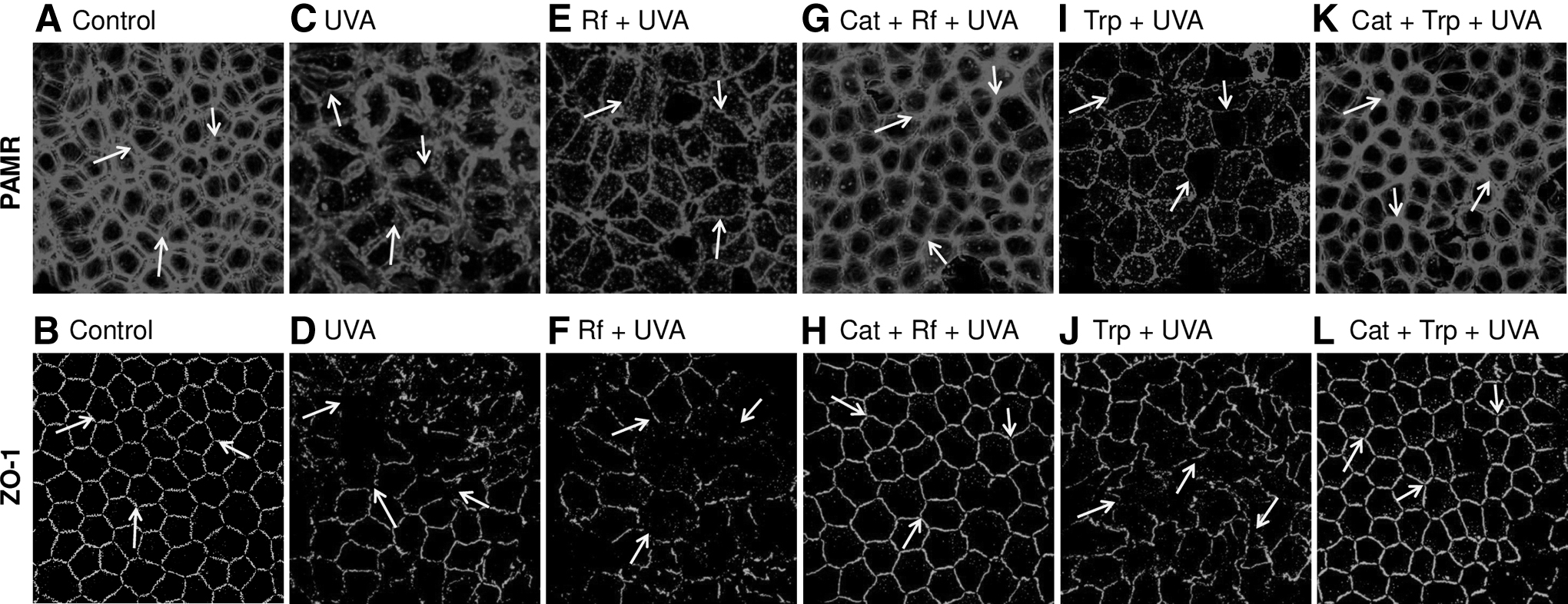
Effect of photodynamic exposure on PAMR and ZO-1. The endothelial surface was exposed to Rf (50 μM, 5 min) or Trp (100 μM, 5 min) and then exposed to UVA for 15 min with and without catalase (10,000 U/mL). Following the treatments, the endothelium was immunostained for ZO-1 and PAMR.
Accumulation of ROS
To confirm the extent of oxidative stress during the photodynamic stimuli, we quantified ROS generation by DCF-DA assay. When cultured endothelial monolayers were exposed to Rf+UVA or Trp+UVA, the green fluorescence of DCF increased significantly (Fig. 4; P < 0.05). The increase is comparable with the fluorescence response following exposure to H2O2 (100 μM) and tBHP (100 μM) (Fig. 4). Furthermore, although accumulation of ROS in response to H2O2 and tBHP treatment is abolished by pre- and co-treatment with SB-203580, the inclusion of catalase in the bathing medium nearly abolished the response to photodynamic stimuli (P < 0.05; Fig. 4). Although the photodynamic stimulus may induce singlet oxygen by type-II reactions, the type-I reaction-mediated release of H2O2 plays a significant role. 24 Thus, as noted in Fig. 3, the damage to PAMR and ZO-1 is blocked by including catalase in the bathing medium before the stress.

Effect of oxidants and photodynamic stimuli on intracellular ROS. Cultured endothelial cells were exposed to oxidants (H2O2 or tBHP) or photodynamic stimuli (i.e., Rf+UVA or Trp+UVA). An increase in the green fluorescence of dichlorodihydrofluorescein indicates elevated ROS. Rf+UVA or Trp+UVA induced ROS comparable with that produced by exposure to H2O2 (100 μM) or tBHP (100 μM). Although ROS accumulation with H2O2 or tBHP is abolished by pre- and co-treatment with SB-203580 (SB), the response to photodynamic stimuli is nearly abolished by catalase (Cat). The data are a compilation of 3 independent trials. P-values are shown above the square brackets, P < 0.05 was considered statistically significant. ROS, reactive oxygen species.
Impact of oxidative stress on MMP
Many cell types show oxidative stress-induced mitochondrial dysfunction and vice versa. 32 The dysfunctional mitochondria can be characterized by altered MMP. Thus, we measured MMP in cultured cells exposed to oxidative stress. Cultured cells responded to H2O2, tBHP, or photodynamic stimuli (i.e., Rf+UVA or Trp+UVA) by a decrease in red/green fluorescence of JC-1, indicating a depolarization of MMP (Fig. 5A, B; P < 0.05). However, the pre- and co-treatment with SB-203580 and catalase blocked the MMP depolarization (Fig. 5A, B; P < 0.05).

Effect of oxidants and photodynamic stimuli on MMP. Cultured endothelial cells were exposed to oxidants (H2O2 or tBHP) or photodynamic stimuli (Rf+UVA or Trp+UVA). Depolarization of MMP was assayed by using JC-1 dye, which is indicated by a decrease in red/green fluorescence.
Impact of oxidative stress on lipid peroxidation
In general, lipid peroxidation is a hallmark ROS accumulation. 33 Thus, we quantified lipid peroxidation in the corneal endothelium ex vivo after exposure to H2O2, tBHP, or photodynamic stimuli (Rf+UVA or Trp+UVA). As given in Fig. 6A, H2O2 and tBHP increased lipid peroxidation (P < 0.05). However, the responses were inhibited by SB-203580. Similarly, increased lipid peroxidation in response to photosensitizers (Rf+UVA or Trp+UVA) were blocked by catalase in the bathing medium (P < 0.05; Fig. 6B).
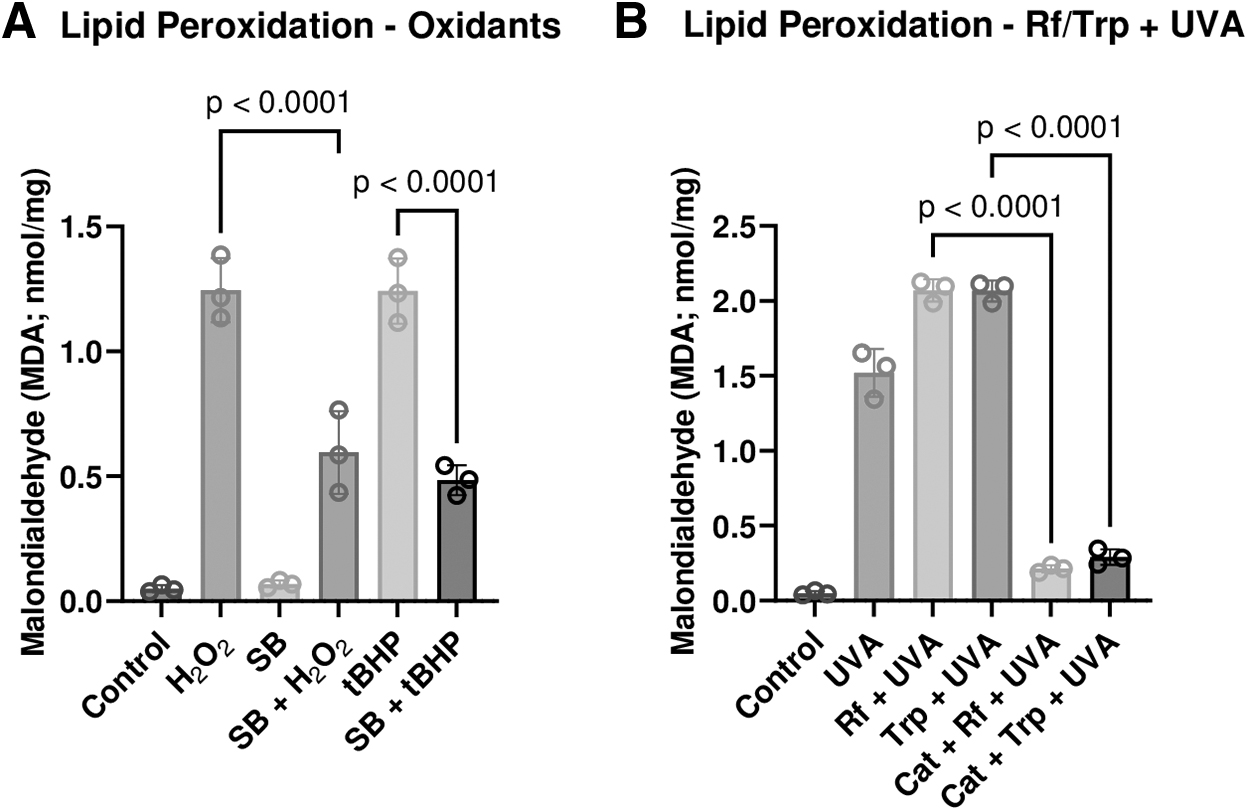
Effect of oxidants and photodynamic stimuli on lipid peroxidation. The endothelial surface was exposed to oxidants (H2O2 or tBHP) or photodynamic stimuli (Rf+UVA or Trp+UVA). The extent of lipid peroxidation is expressed as the amount of malondialdehyde released from the endothelial cells.
Impact of oxidative stress on the release of LDH
LDH release is indicative of damage to the plasma membrane secondary to oxidative stress. Accordingly, we examined the response with endothelium in corneas ex vivo. 34 As given in Fig. 7A and B, H2O2, tBHP, or photodynamic stimuli (Rf+UVA or Trp+UVA) increased %LDH release. However, the release was inhibited by pre- and co-treatment with SB-203580 (P < 0.05; Fig. 7A). %LDH release was also 2-fold higher in the presence of photosensitizers Rf/Trp+UVA treatment compared with UVA alone (P < 0.05; Fig. 7B). However, %LDH release was reduced by including catalase in the bathing medium during the photodynamic treatment (Fig. 7B; P < 0.05).
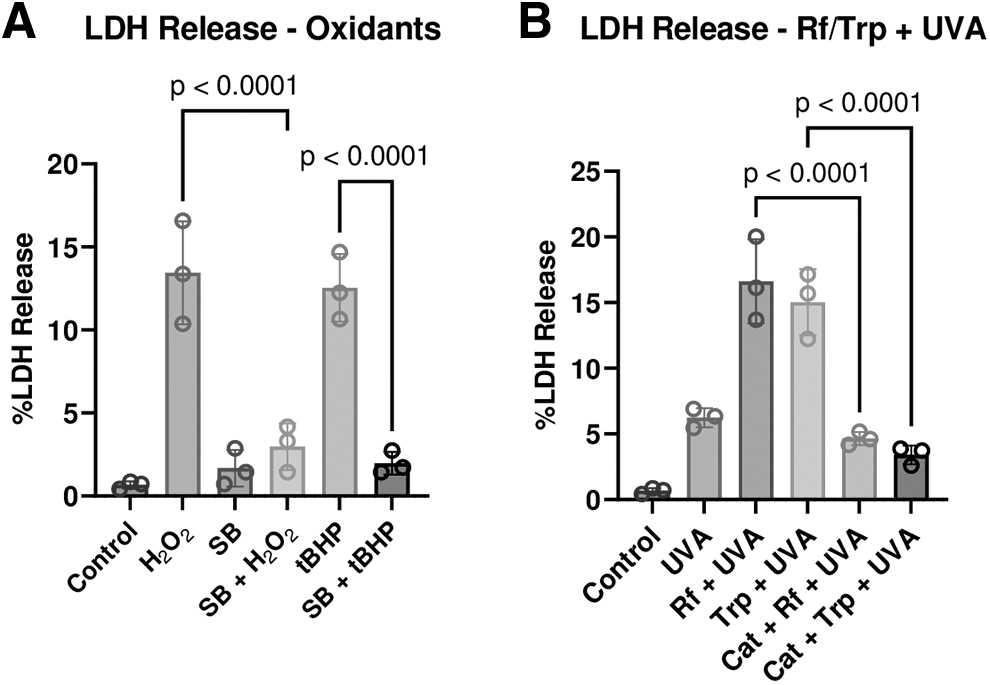
Effect of oxidants and photodynamic stimuli on %LDH release. The endothelial surface was exposed to oxidative stress (H2O2 or tBHP) or photodynamic stimuli (Rf+UVA or Trp+UVA). The %LDH release was 15.4 ± 0.7% (n = 3) in response to Triton X-100 exposure (0.2% for 1 h).
Impact of oxidative stress on the permeability of FITC-avidin
We examined the loss of barrier function by quantifying the transport of FITC-avidin across cultured endothelial monolayers. 25 As a macromolecule, FITC-avidin does not cross the intact monolayers (Fig. 8A, B). However, exposure to UVA alone resulted in a noticeable flux across the monolayer (Fig. 8G, H). Exposure to H2O2 or Rf+ UVA resulted to a significant increase in the flux of FITC-Avidin, and subsequent binding to the gelatin-biotin complex at the basal surface. This is evident by the increase in fluorescence, especially at cell-cell junctions (Fig. 8D, J, C, and I; Fig. 9; p < 0.0001). Pre and co-treatment of SB-203580 (Fig. 8E, F; p < 0.05) reduced the FITC fluorescence. Similarly, the inclusion of catalase blocked FITC-Avidin penetration across the monolayer, as shown in Fig. 8K, L and Fig. 9 (p < 0.0001).
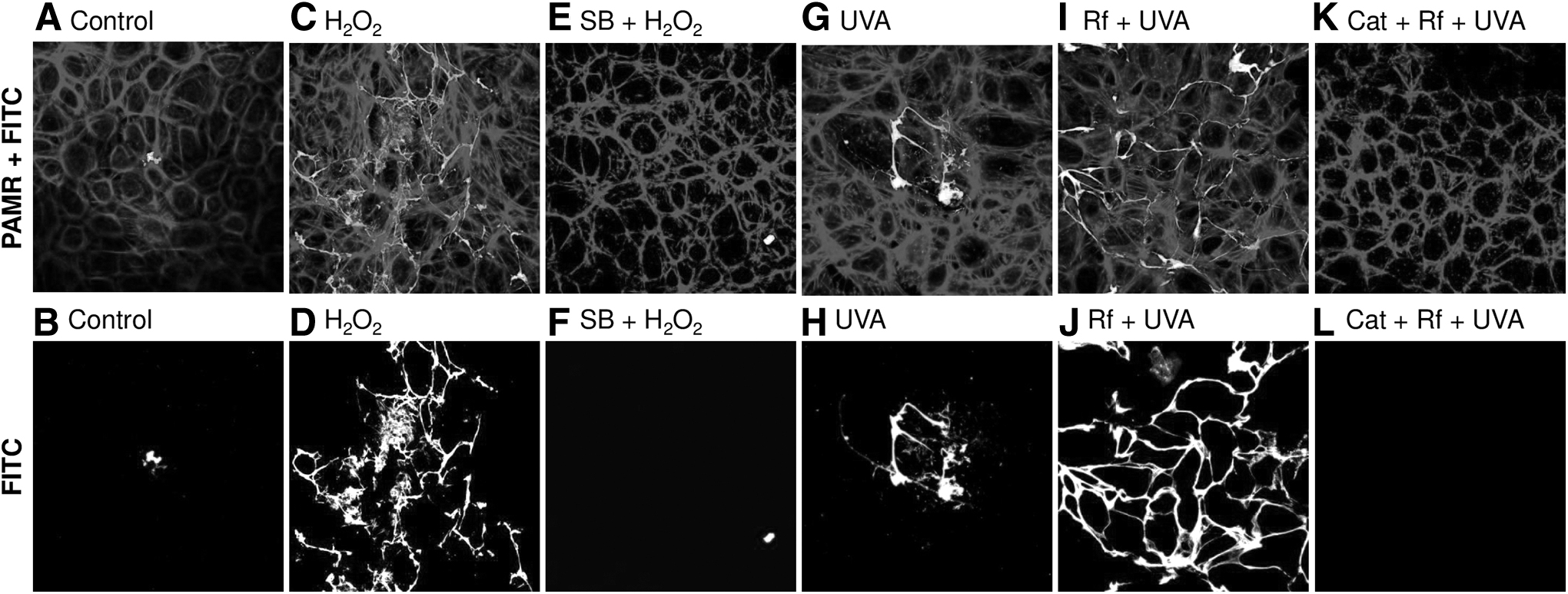
Effect of oxidants and photodynamic stimuli on endothelial barrier function. Cultured endothelial cells grown to confluence on biotinylated gelatin-coated coverslips were exposed to H2O2 or Rf+UVA with or without pre- and co-treatment with SB-203580 (SB) or catalase (Cat). Following treatments, cells were immunostained for PAMR and imaged along with FITC.
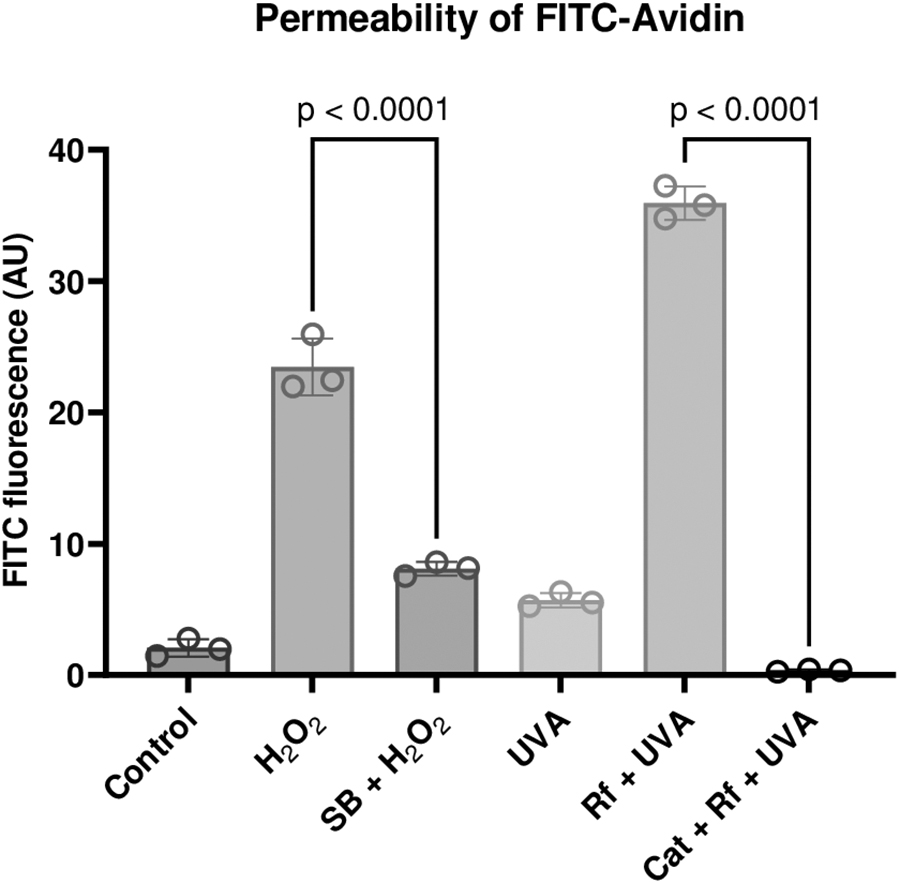
Effect of oxidants and photodynamic stimuli on permeability of FITC-avidin. Following experiments as in Fig. 8, the fluorescence intensity of FITC was quantified using ImageJ. The increase in FITC-avidin indicated elevated permeability and breakdown of the barrier function. The flux is shown to be for H2O2 and Rf+UVA compared with the pre- and co-treatment of SB-203580 or catalase. The data are a compilation of 3 independent trials. P-values are shown above the square brackets, and P < 0.05 was considered statistically significant.
Overall, elevated ROS, depolarization of MMP, increased %LDH release, and pronounced lipid peroxidation confirm oxidative stress as intended by exposure to oxidants and photodynamic stress. The impact of the stress on the AJC is demonstrated by the destruction of PAMR and loss of barrier integrity. The latter is also evident in the increased permeability to FITC-avidin.
Discussion
The primary symptom in the early stages of FECD is blurred vision, which is experienced in the early hours of the morning.1,4 It is attributed to persistent stromal edema that prevails when eyes are closed during sleep. 4 As an insidious disorder, the problem of blurred vision sustains longer, becoming more pronounced with the continued decline in ECD and coalescence of guttae. 35 Although the advanced stage of the disorder is treated by endothelial or full-thickness keratoplasty, there is no pharmacological approach to treat the symptoms. 36 Therefore, developing topical drugs is imperative for effective clinical management of the disorder in the early stages. The stromal edema, in principle, can be attributed to the loss of ECD and the loss of barrier and pump functions. 37 Thus, we have examined whether the hallmark of the disease, namely oxidative stress, 3 affects the PAMR and thereby subverts the barrier function.
This study is a sequel to our recent study in which acute oxidative stress was shown to break down the microtubules in the endothelium, leading to the destruction of PAMR and ZO-1 via p38 MAP kinase activation. 24 In this study, we have extended the investigation into the porcine endothelium associated with intact tissue ex vivo.
Our first experiments demonstrate the destruction of AJC via the RhoA-Rho kinase axis (Fig. 1). Specifically, we induced a breakdown of AJs by removing extracellular Ca2+ to evoke activation of Rho kinase. 12 The latter induces contraction of the PAMR, pulling apart the tight junctions (Fig. 1C, D). We also show the normalization of PAMR and resealing of tight junctions following the reformation of AJs by Ca2+ add-back (Fig. 1E, F). Thus, this experiment established the application of immunocytochemistry to demonstrate the tight coupling between PAMR and the barrier integrity, similar to our previous work based on the effect of histamine, 13 thrombin, 13 and nocodazole 20 on phosphorylation of myosin light chain and trans-endothelial electrical resistance. These experiments were mostly carried with bovine-cultured endothelial monolayers.
As demonstrated previously, we used two approaches to elicit acute oxidative stress on endothelial cells. In the first method, we exposed the endothelial surface associated with intact corneal buttons to membrane-permeable oxidants: H2O2 and tBHP. Both agents damaged PAMR and tight junctions (Fig. 2C, D, I, J), similar to observations with cultured cells. 24 Pre- and co-treatment with SB-203580 reduced the effects of the oxidants (Fig. 2G, H, K, L), confirming that they elicit activation of p38 MAP kinase. In the second method, the endothelial layer was exposed to UVA with Rf/Trp as photosensitizers. Both agents significantly destroyed the PAMR and ZO-1 (Fig. 3E, F, I, J). We attribute these effects to H2O2 released by the type-I photodynamic reaction. This is because catalase, which decomposes H2O2, prevented the response to photodynamic stimuli (Fig. 3G, H, K, L). 24 These results also agree with our recent findings with cultured cells. 24
Figures 4–7 demonstrate the relative extent of oxidative stress induced by the photodynamic stimuli compared with the known oxidants (i.e., H2O2 and tBHP). Accordingly, we note that the photodynamic stimuli induce ROS accumulation comparable with the membrane permeable oxidants (Fig. 4).
These observations confirm that type-I reactions that culminate in the production of H2O2 underlie the damage to the cytoskeleton and AJC in response to the photodynamic stress. On the contrary, type-II reactions, which mediate the release of singlet oxygen, are likely to induce rapid cell death but are unlikely to have played a role in elevated intracellular ROS because catalase nearly abolished the increase in DCF fluorescence. On the contrary, as noted elsewhere, the type-I reactions result in H2O2. 24 The latter being permeable across plasma membrane results in cytoskeletal damage and AJC destruction, as given in Fig. 2. Finally, similar to H2O2 and tBHP, we also note that photodynamic stimuli resulted in depolarization of MMP (Fig. 5A, B), enhanced lipid peroxidation (Fig. 6A, B), and increased %LDH release (Fig. 7A, B). Finally, an increased paracellular flux of FITC-avidin was observed in response to the photodynamic stimuli similar to direct exposure to oxidants (i.e., H2O2) (Figs. 8 and 9).
Taken together, we have reconfirmed a versatile photodynamic approach to induce regulated oxidative stress on endothelium associated with intact corneal buttons. The protocol is amenable for use in drug discovery studies in the context of FECD and other situations where the endothelium is known to sustain oxidative stress. For example, recently, we have demonstrated that cold storage of donor corneas induces a loss in the barrier function via oxidative stress involving mechanisms similar to those described for response to photodynamic stimuli. 15 Moreover, cytokines also induce stress signaling downstream of oxidative stress.10,14
Conclusions
Acute oxidative stress breaks down PAMR and ZO-1 via p38 MAP kinase-dependent mechanisms in the corneal endothelium. The efficacy of catalase in opposing the damage to AJC confirms that H2O2 released via type-I reactions underlie responses to the photodynamic stress. Thus, the photodynamic protocol we have tested can be an alternative to investigate oxidative stress in the endothelium for unraveling the pathophysiology of FECD. The protocol can also be used for drug discovery against loss of barrier function in response to oxidative stress in the corneal endothelium.
Footnotes
Authors' Contributions
C.A.: Methodology, validation, formal analysis, investigation, writing, visualization. A.S.: Methodology, supervision, validation, formal analysis. S.H.R.: Resources, supervision, project administration, funding acquisition, writing, validation. S.P.S.: Conceptualization, methodology, validation, formal analysis, writing, visualization, supervision.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work is supported by the Science and Engineering Research Board-Extramural Research grant (EMR/2017/002449) from Department of Science & Technology, Government of India, and Roche Collaborative Research Fellowship from Association for Research in Vision Science and Ophthalmology (United States) to Sudhir H. Ranganath and in part by the intramural research grant from Sree Siddaganga Education Society, Tumakuru (3150/1516).