
Abstract
Abstract
Background:
Medication registration currently requires evidence of safety and efficacy from adequately powered phase 3 studies. Pharmacovigilance (phase 4 studies, postmarketing data, adverse drug reaction reporting) provide data on more widespread and longer term use. Historically, voluntary reporting systems for pharmacovigilance have had low reporting rates, relying on ad hoc reporting and retrospective chart reviews, or prospective registries have often been limited to specific drugs or clinical conditions. Furthermore, these data are often irrelevant in hospice and palliative care due to the timeliness of which such data become available and the unique characteristics of our population and prescribing: compounding comorbidities, progressive organ failure, accumulation of symptom-specific medications, tendency to attribute toxicity to disease progression, use of old, off-patent medications, and incorporation of evolving evidence. There is a need for prospective, systematic pharmacovigilance in hospice and palliative care.
Method:
Here we describe an international, Web-based, 128-bit secure initiative to collect pharmacovigilance data documenting net clinical benefit and safety of common medications. The intention is for a diverse and large group of clinical units to record data prospectively on a small deidentified consecutive cohort of patients started on the medication of interest. A new medication would be studied every 3 months. Three key time points (different for each medication) will be assessed for each patient, collecting easily codefiable data at baseline, a point at which clinical benefit should be experienced, and a point at which short- to medium-term toxicities may occur. Toxicities can additionally be recorded at any time they occur. Data collection will take a maximum of 10 minutes per patient.
Conclusion:
The intention is to create an efficient, relevant system to improve hospice and palliative care with maximally generalizable results.
Introduction
These same agencies are starting to put a greater emphasis on longer term outcomes and safety. Globally, regulatory agencies are looking toward pharmacovigilance studies to demonstrate long-term safety and effectiveness. Studies and efforts contributing towards this goal have many labels, including; phase 4, pharmacovigilance, postmarketing surveillance, safety monitoring, late-phase data, real-world data, and postregulatory registries. These studies may or may not be in response to regulatory requirements.
The evidence base for the clinical use of medicines is complex and continually evolving, even for well-established medications in widespread use (most of which are now off-patent). As treatments change, and the prevalence and combinations of comorbid illnesses evolve, the relative safety and efficacy of medicines also change. To provide the evidence base for evolving clinical care, processes are needed to systematically monitor and quantify common toxicities, rare effects, long-term and cumulative effects, and interactions with new medications introduced into practice Few registration studies are powered on safety outcomes. Efficacy or effectiveness are the usual end points for power calculations. The harm that a medication can cause is often not detectable due to lack of power because of event rates, time to events, or the large numbers of random events collected as safety outcomes in clinical studies.
Current processes for postmarketing surveillance have substantial limitations. Voluntary pharmacovigilance programs suffer from low reporting rates in all clinical disciplines even when events are directly attributable to a drug–host or drug–drug interactions.
1
Industry pharmacovigilance programmes suffer from being focused on a single medication and the inherent conflicts between the interests of shareholders and patient safety. Clinical practice databases, such as the UK General Practice database, are increasingly the source of information about drug harm, but have inherent limitations as the data are collected for other purposes and key information may be missing.
2
Data available in the public domain rely on:
1. Ad hoc reporting 2. Retrospective case note audits (i.e., chart reviews): or 3. Prospective audits where there is an agreed systematic set of data to be recorded (i.e., registries).
Ad hoc reporting of adverse events is inconsistent and rare. Retrospective chart reviews or local audit is often inconsistently conducted between sites, limiting ability to combine information to determine large-scale patterns of benefit and harm. Prospective registries provide great benefit, but historically have been expensive and cumbersome to conduct.
New technologies can help in the latter model either by more efficient use of existing datasets or through collection simultaneously across multiple sites. The merging of large administrative datasets may allow the testing of hypotheses concerning adverse events. 3 Many aggregated datasets for comparative effectiveness research, including registries and electronic health record (EHR) data, are now being used to monitor more systematically for longitudinal drug effectiveness and safety including adverse event reporting.
In hospice and palliative care, longitudinal pharmacovigilance data have not been systematically collected. Clinicians and patients may attribute medication-related toxicity to disease progression, or miss clinically relevant toxicities simply because they were not actively sought. The culture of audit and safety reporting is scant, with the use of large scale administrative datasets and EHRs uncommon. Hospice and palliative care needs to develop and embed the culture and approaches for good pharmacovigilance especially given that symptom-specific medications are added to medications for long-term comorbid conditions.
A Case for Pharmacovigilance in Hospice and Palliative Care
Phase 4 pharmacovigilance data directly complement phase 3 studies. Both elements are needed if the evidence base for hospice and palliative care practice is to continue to evolve to improve patient outcomes. 4
In hospice and palliative care, the evidence base supporting application of interventions in clinical practice is complex and continually evolving. Interventions are diverse, and frequently coadministered. Medications used are often off-patent, therefore, manufacturer-sponsored and regulation-required safety monitoring is unlikely. The population encountered in palliative care practice is also evolving, with widening profiles of diseases, multimorbidity, chronicity, and physiologic changes. Systematic monitoring for commonly encountered toxicities, rare effects, long-term and cumulative effects, and interactions with current and new medications as they become available is rare.
The hospice and palliative care patient population is characterized by increasing frailty, a progressive catabolic state, varying degrees of progressive organ system impairment, and the need to add symptom control medications while simultaneously managing long-term comorbidities. 5 The hospice and palliative care population is probably the subgroup in clinical practice at the greatest risk of iatrogenic harm given that this risk increases greatly with the number of medications. 6
There is therefore a need to evaluate repeatedly and systematically the performance of medications in the target audience for whom they are prescribed. Although, for most medications the numbers needed to treat (NNT) and to harm (NNH) may be defined by a pretreatment probability through clinical trials for short-term scenarios, both parameters require longer term data beyond a primary efficacy end point. Furthermore, pretreatment event probability will vary increasingly later in the palliative care disease trajectory. For all medications, the relationship between NNT and NNH is dynamic and will continue to be adjusted as phase 4 data become available, and the palliative population and the medications they are prescribed evolves. NNT and NNH assume a linear relationship between events and the initiation of therapy, 7 but both parameters will change for individual patients as their conditions change.
Not only do hospice and palliative care practitioners need to know when to initiate a medication but constantly updated evidence to inform when and how to discontinue medications is also required.
7
Any clinical intervention should have an evaluable end point with the time to that end point identified at the time of initiating treatment.
8
Likewise, the time course of major, clinically relevant harms should be ascertainable from the literature. At their most basic, outcomes will include predicted benefit being greater than the predicted harm or vice versa. Medications may be continued or changed by:
••Ceasing the medication; ••Dose reduction; ••Substituting a similar medication that has less adverse effects; or Adding medications to manage the adverse effects.
Adding medications specifically to manage the side effects of another medication can potentially lead to “therapeutic momentum” or a “prescribing cascade” where more and more medications are added to treat problems from existing medications.9,10
A key element is to be able to attribute benefits and harms to a specific medication. Bradford-Hill 11 proposed a series of criteria for assessing causality in populations that has been adapted to pharmacovigilance. Naranjo et al. 12 proposed a series of theoretical and practical criteria for assessing causality of harm of medications in individuals including rechallenge. In practice, severity, scientific plausibility, and the temporal relationship between a medication starting and an effect being experienced may be enough to ensure that the person is not rechallenged with the drug in routine clinical practice especially in hospice and palliative care. Of the nine criteria proposed by Naranjo and colleagues to attribute causality to a medication for an adverse event, five will be used in the clinical assessment to aid in understanding likely attribution of a relationship between the medication and the observed side effect (Table 1). Those that will be omitted are not realistic for routine use in end-of-life care.
The ability to perform pharmacovigilance studies in hospice and palliative care is becoming more practical. New and emerging information systems allow the rapid acquisition of data to inform practice in a way that has not been available before. 13 Multisite collection with minimal impost on clinicians and no impost to patients allow a range of specified data to be collected prospectively to ensure that the evidence base for safety continues to evolve rapidly. 14
A New International Collaborative Pharmacovigilance Program
The aim of this collaborative program is to systematically collect data on the clinical benefits and toxicities of medications used for symptom control in hospice and palliative care practice at a level that is consistent with and reflective of contemporary clinical practice, with formalized data collection embedded within routine care. The intention is to create a relevant, efficient system to document net clinical benefit and safety of common medications used in hospice and palliative care. By creating a simple and internationally accessible system, a diverse and large group of clinical units can participate.
A new medication would be studied every 3 months. Three key time points (different for each medication) will be assessed for each patient, collecting easily codefiable data at baseline, a point at which clinical benefit should be experienced, and a point at which short- to medium-term toxicities may occur (Fig. 1). Toxicities can additionally be recorded at any time they occur. Data collection will take a maximum of 10 minutes per patient. Medication/symptom dyads in the first 12 months will include metoclopramide/nausea, haloperidol/delirium, dexamethasone/appetite, and gabapentin/neuropathic pain.
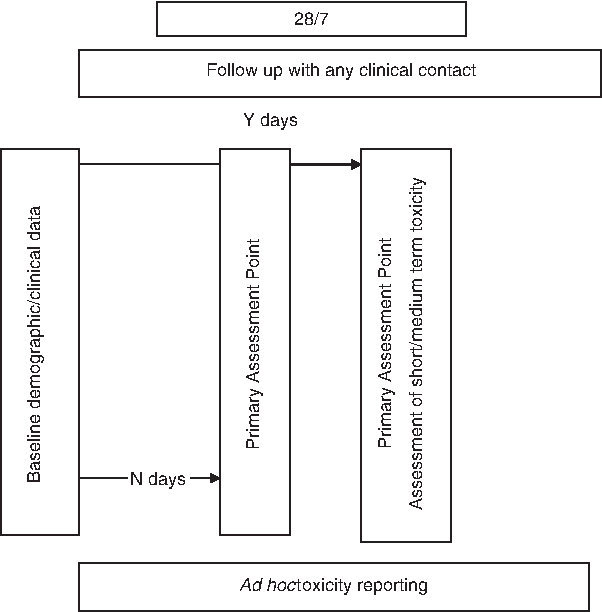
Generic representation of time points for the multisite prospective pharmacovigilance data collection.
Specific elements of this pharmacovigilance project include:
• A simple registry format, whereby a consecutive cohort of people internationally, newly started on the medication of interest have data collected prospectively against an agreed pro forma (whether as an inpatient, outpatient or in the community); • A series of medication evaluations with a new medication/symptom dyad commencing every 3 months. (If a medication has more than one clinical indication, each indication will be studied at a separate time. For example, haloperidol may be used for nausea, delirium and sedation; each 3-month study would look at only one clinical indication); • set time points including:
(i) baseline; (ii) a single time point at which clinical benefit will be evaluated; and (iii) a time point for toxicity assessment.
Ad hoc time points will include withdrawal at any time and any time the person has toxicity that comes to the attention of the clinical team and oversight by an audit development committee with expertise including palliative medicine, clinical pharmacology, and pharmacy.
Data are kept to a minimum with no identifying information collected. By choosing a small dataset, the burden on individual clinicians will also be minimized. For example, although gender and age will be collected, date of birth will not be collected, ensuring that not only are these deidentified data, they will also be un-reidentifiable. Comorbidities will be assessed using the Charlson comorbidity Index 15 and functional status will be assessed using the Australian-modified Karnofsky Performance Scale. 16 Clinical data collected are intended to be consistent with information that is already collected as a part of the normal process of prescribing a new medication.
Data will be collected on a 128-bit secure website (www.caresearch.com.au) that will be customized for each new dyad of study medication and the specific palliative clinical indication being studied. This will allow real-time collation of the data for rapid reporting with the aim that each medication/indication will be reported in the Journal of Palliative Medicine.
Since data will be reported external to an organization, and this program cannot be considered as quality monitoring only, some notification of human subjects authorities is required. However, since the data collected are a part of standard of care, it is anticipated that most hospice and palliative care programs will be able to participate through a written waiver from the Research Ethics Committee/Institutional Review Board or, at worst, as a low-risk application that can be expedited. Written patient consent should not be required.
Many countries have national authorities or credentialing bodies that seek demonstration of longitudinal assessment of practice and quality monitoring as a part of documented best practice (e.g., Joint Commission in the United States). 17 Participation in this pharmacovigilance initiative can contribute to such processes. Also, the data elements can be integrated into other standard data collection activities, such as routine quality monitoring (e.g., PCOC, QDACT) or EHRs, and these data can be transferred to the pharmacovigilance program for inclusion in rapid reporting of results.13,18
This program will allow a wide range of practices and practitioners from around the world to participate in this process allowing rapid updating of information. This will improve generalizability of findings. The approach is intentionally efficient, inexpensive, and standardized, directly responding to the problems encountered in wide-scale pharmacovigilance programs in the past. Individual practitioners will be able to reflect on their current practice in the light of toxicities that are specifically sought, with reports requiring less than 10 minutes to complete. For the discipline, it will provide another evidence stream to inform net clinical benefit, not simply a view of who has responded to therapy.
This mechanism provides an exciting opportunity for an international collaboration to improve clinical practice. Because of the focus on 3-month time periods, in its current configuration this surveillance program does not address the long-term aspects of pharmacovigilance—cumulative effects, nor long-term toxicity. It will also be limited at this time in its ability to identify specific drug–drug interactions, but these are potential future directions if the collaborative is successful.
Conclusion
There is an urgent need to create a network of hospice and palliative care programmes committed to improving directly the quality of care that we offer to patients and their families.
By a large number of services committing to collect data over a 3-month period on a small number of consecutive patients newly introduced to a new medication for a medication/indication dyad, a large amount of efficacy and toxicity data will be available in a way that has not been possible before in hospice and palliative care. Ultimately, patient-centered care is about providing patients with sufficient information as to whether or not they would consider using a particular therapy.
Any clinical group can join this initiative. Please contact the corresponding author david.currow@health.sa.gov.au
Footnotes
Author Disclosure Statement
No competing financial interests exist.