
Abstract
Background:
This study investigated the efficacy and safety of a combined oral contraceptive (COC) containing estradiol valerate/dienogest (E2V/DNG).
Methods:
This was a multicenter, noncomparative, 13-cycle (extended to 28 cycles) study conducted in the United States and Canada. Contraceptive efficacy was calculated as a Pearl Index for 13 cycles, based on all on-treatment pregnancies; bleeding patterns were calculated based on bleeding and spotting information recorded daily in diary cards. Safety events during a 16-month extension study were added to the 1-year data.
Results:
In total, 499 women, aged 18–35 years, were enrolled, and 490 of them were included in the full analysis set for contraceptive efficacy. Five pregnancies occurred in the first year (unadjusted Pearl Index=1.64). In cycles 1–12, an average 23.5% of women had absent scheduled (withdrawal) bleeding. Among women with scheduled (withdrawal) bleeding, bleeding started after a median of 2 days after intake of the last DNG-containing pill. For safety, data included from 147 women followed over an additional 16 months were added to the original 13-cycle data set. Treatment-related adverse events (AEs) occurred in 51.8% of women; 14.9% discontinued because of AEs over the entire 28-month study period.
Conclusion:
A COC with E2V and DNG was shown to provide effective contraception in women aged 18–35 years in North America.
Introduction
S
The first estrogen used in COCs was the prodrug mestranol, which was replaced by its potent, active form, ethinyl estradiol (EE). Over the years, attempts have been made to replace EE with estradiol (E2)-based compounds. Compared with EE-based formulations, E2-based COC formulations have fewer hepatic and hemostatic effects. 1 Although earlier attempts to develop monophasic and biphasic E2-based COCs yielded promising results in terms of ovulation inhibition and contraceptive efficacy, 2 –6 these formulations were associated with unacceptably high rates of bleeding irregularities 3 –6 and bleeding-related discontinuation.
With the development of progestins with stronger endometrial effects (i.e., progestins with high progestational potency in transactivation assays), 7,8 the use of such compounds as 17β-estradiol in COCs has been more successful. In 2010, the U.S. Food and Drug Administration (FDA) approved a COC formulation that contained estradiol valerate (E2V) and dienogest (DNG) in a dosing regimen that was designed to minimize cycle control-related issues observed with previously studied E2-based monophasic and biphasic COC formulations. A second E2-containing monophasic formulation has been approved by the European authorities.
E2V is an ester of the natural human 17β-estradiol, 9 with pharmacodynamics and pharmacokinetics similar to orally administered micronized E2. 10,11 DNG is a progestin that displays a pharmacodynamic profile that combines the characteristics of both 19-norprogestins and progesterone derivatives. 12,13 DNG binds the progesterone receptor with high specificity but with low relative affinity compared with progesterone. In addition, DNG is known to have a pronounced progestational activity on the endometrium, effectively inhibits ovulation with low antigonadotropic activity, and lacks estrogenic, antiestrogenic, and androgenic activities. 13 A notable attribute is that a relatively high fraction (approximately 10%) of DNG is free in the serum, and 90% is bound nonspecifically to albumin, with no or minimal affinity to sex hormone–binding globulin. 12
The approved regimen of E2V/DNG, administered using an estrogen step-down and progestin step-up, was identified in clinical trials as having the lowest effective dose of DNG in combination with E2V that achieved a high degree of ovulation inhibition and good cycle control. 14 The E2V/DNG dose regimen was designed specifically to provide early estrogenic dominance so as to allow for limited initial endometrial proliferation and for endometrial stroma stability during the progestin-dominated mid-to-late part of the cycle, thought to optimize cycle control. This formulation was first approved for the prevention of pregnancy in Europe in 2009, based on the results of two multicenter trials conducted in Europe 15 and North America. The results of the North American study, which evaluated the contraceptive efficacy and safety as well as the bleeding profile of E2V/DNG, are presented here.
Materials and Methods
Study design and participants
This multicenter, open-label, noncomparative study evaluated the efficacy and safety of E2V/DNG (Natazia®, Bayer HealthCare Inc. Wayne, NJ [named Qlaira® in Europe]). The study planned to enroll 480 subjects in order to have 400 subjects complete 13 cycles (28-day cycles) of treatment. In order to obtain additional, longer-term information about the efficacy and safety of E2V/DNG, the study was extended to include the experience of women who received treatment for a maximum of 28 cycles. Eligibility for enrollment into the extension phase of the study was based on demonstrated compliance with study medication during the initial planned study phase (i.e., only those active subjects who, at the end of the 13-cycle study period, had not reported the use of back-up contraception because of missed tablets for more than 2 cycles, were eligible). The study (protocol number: 304742;
The trial recruited healthy women aged 18–35 years seeking contraception. Smokers over the age of 30 years, as well as women with a body mass index (BMI) of more than 30 kg/m2, were not included in the study. Other exclusion criteria for the trial were consistent with the contraindications and special warnings and precautions for COC use included in the labels of COCs containing EE. Briefly, these exclusion criteria included pregnancy or lactation in the past 3 months and history or presence of any of the following conditions: thromboembolic disease, chronic inflammatory bowel disease, hemolytic uremic syndrome, pancreatitis, diabetes, sickle cell anemia, depression, drug abuse, or alcoholism. Current active problems with undiagnosed abnormal vaginal bleeding, thyroid disorders, hepatic disease, and known or suspected premalignant or malignant diseases were also exclusionary.
Each woman's history was obtained at a screening visit. Medical and surgical history focused on events within the 6-month period prior to study enrollment. Obstetric history included data on pregnancies, births, and abortions. Menstrual history included information on menarche and recent bleeding patterns. Additionally, physical and gynecological examinations, including breast examination by palpation and cervical cytology tests, were performed. Daily diary cards were used to track compliance with treatment as well as to assess bleeding patterns.
Study treatment
The birth control pill formulation studied comprised E2V 3 mg on days 1 and 2, E2V 2 mg/DNG 2 mg on days 3–7, E2V 2 mg/DNG 3 mg on days 8–24, E2V 1 mg on days 25 and 26, and placebo on days 27 and 28 (Fig. 1).

A 28-day regimen of estradiol valerate (E2V)/dienogest (DNG) incorporating stepped-down doses of E2V and stepped-up doses of DNG over 26 days of active treatment.
In the first treatment cycle, the first tablet was taken on the first day of menstrual bleeding in new COC starters or on the first day of scheduled (withdrawal) bleeding in women switching from another combined hormonal method. The subjects were allowed to take the tablets either in the morning or the evening; the interval between any two tablets was to be as close as possible to 24 hours.
Contraceptive efficacy
The primary efficacy variable was the number of observed pregnancies during study treatment. For the purposes of this article, contraceptive efficacy is represented using the Pearl Index (number of pregnancies per 100 woman-years of exposure). The Pearl Index and corresponding upper 95% confidence interval (CI) were calculated as an unadjusted Pearl Index, using data obtained during the first 13 cycles of treatment. As required by the FDA, the Pearl Index calculation was based on all on-treatment pregnancies and all pregnancies that occurred in the first 7 days after discontinuation of study medication (i.e., within 1 week of the last placebo pill) in women aged 18–35 years.
At each study visit, investigators collected information about the use of any additional nonhormonal contraceptive methods by cycle so that only at-risk cycles were included in the analysis. For each pregnancy, the local investigator was required to document, insofar as possible, the calculated date of conception, the measures used to estimate that date, and the pregnancy outcomes. To categorize the failures observed, the investigator was also required to ascertain whether the subject had taken any substance or had had a concomitant illness (e.g., vomiting or diarrhea) that might have affected pill absorption or whether there were any tablet-use errors that may have contributed to the pregnancies.
Bleeding outcomes
Each subject recorded bleeding on a daily basis, using diary cards. “Bleeding” was defined as vaginal bleeding the subject categorized as light, normal, or heavy, that, based on her personal experience, required the use of sanitary protection. “Spotting” was defined as vaginal blood in an amount that did not require the use of sanitary protection other than panty liner(s).
Bleeding episodes were identified and analyzed, using data from the diary cards, as scheduled (withdrawal) bleeding and unscheduled (intracyclic). A scheduled (withdrawal) bleeding episode under treatment was defined as the first bleeding that started no earlier than day 21 (i.e., not more than 4 days before DNG was withdrawn) and continued without interruption. Both the onset and the duration of bleeding were assessed. A cycle was counted as absence of scheduled bleeding if there was no bleeding or spotting from day 21 of the cycle to day 20 of the subsequent cycle. All other (unexpected) bleeding episodes were reported as unscheduled (intracyclic) bleeding. It should be noted that all bleeding in the first treatment cycle was, by definition, categorized as unscheduled (intracyclic) bleeding, because of the requirement for a first-day start.
In the current article, uterine bleeding outcomes except for scheduled bleeding are reported from cycles 1–13. Scheduled (withdrawal) bleeding is reported for cycles 1–12 only, because the end of diary records occurred on day 28 of cycle 13, before many women would be expected to start or finish their scheduled bleeding for that last cycle.
Safety and tolerability
Safety was assessed by undertaking physical and gynecological examinations (including a cervical cytology test) and by monitoring vital signs, adverse events (AEs), and the use of concomitant medications. An analysis of AEs was performed over the entire study period (i.e., up to 28 cycles) in the full analysis set (defined as all subjects admitted to the treatment phase who took at least one pill of study medication and from whom at least one observation after admission to treatment was available). AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) dictionary. Metrorrhagia (i.e., irregular or intracyclic uterine bleeding) and other similar terms were used as defined in the study protocol. An AE was considered to be treatment-emergent if it occurred any time from the initial administration of the study medication until the end of the study. The relationship of the AE to the study drug was rated by the investigator as “none,” “unlikely,” “possible,” “probable,” or “definite,” using standard FDA-defined criteria.
A subjective assessment of the study treatment was performed using a questionnaire that was administered at the participants' final visit. The questionnaire was designed to capture participants' overall satisfaction with the study treatment (“very satisfied,” “somewhat satisfied,” “neither satisfied nor dissatisfied,” “dissatisfied,” or “very dissatisfied”), their intended choice of contraceptive method in the future (“continue with the study treatment,” “use a different hormonal contraceptive,” “use a different contraceptive method,” “discontinue use of all types of contraceptives,” or “don't know”), and their perception of their current overall physical and emotional well-being comparing end-of-study to baseline (“much better,” “somewhat better,” “the same,” “somewhat worse,” or “much worse”).
Results
Subjects
Of the 583 women who were screened, 499 were enrolled into the study. Overall, 490 women who received E2V/DNG and had at least one post-baseline assessment were included in the full analysis set. The baseline demographic characteristics of these women are shown in Table 1. The racial distribution of women represented had some diversity, and fewer than 1 in 5 women smoked.
Data are shown as mean±standard deviation unless otherwise stated.
In the month before screening (information missing for 9 women).
Of the 490 women included in the full analysis set, 290 (59.2%) completed cycle 13 of the study. There were 12 (2.4%) women for whom study-completion status was unknown during the first 13 cycles of the study. The population in the extension phase of the study consisted of 147 women from the original population who completed all 13 cycles and had been reasonably compliant; that is, they had not reported the use of back-up contraception because of missed tablets for more than 2 cycles, and they had agreed to participate. Figure 2 summarizes the flow of patients through the study. Of the women who discontinued study medication within the first 13 cycles (i.e., 172), 68 (13.9%) did so because of AEs (Fig. 2). The discontinuation rate due to AEs for the combined experience of the original study and the extension study was 14.9%.
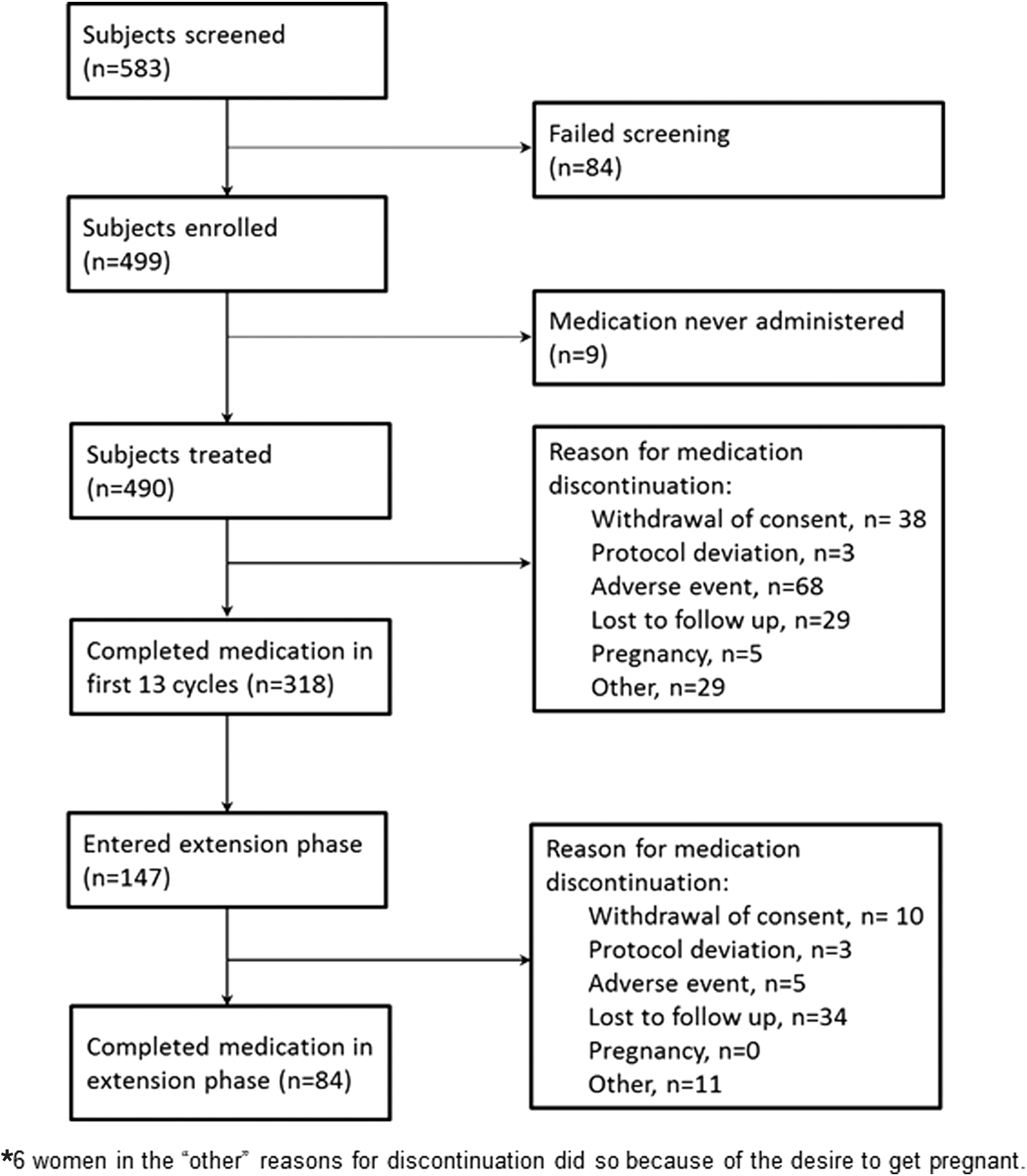
Flow of women through the first 13 cycles by whether they subsequently entered the extension phase of the study.*
Contraceptive efficacy
During the first 13 cycles, five pregnancies occurred over an exposure time of 3,969 cycles, resulting in an unadjusted Pearl Index of 1.64 (upper limit of 95% CI: 3.82). Only pregnancies that occurred while the subjects were still in the treatment phase of the study were used to calculate the Pearl Index. Additional pregnancies that occurred following treatment cessation in the base study included one pregnancy that occurred 9 days after pill cessation (the subject discontinued specifically in order to conceive) and three that were more than 14 days after cessation. In the extension phase of the study, one pregnancy occurred on treatment, and a second occurred more than 14 days after pill cessation.
Cycle control and bleeding outcomes
Scheduled (withdrawal) bleeding
The percentage of women who had scheduled (withdrawal) bleeding fluctuated slightly, averaging 76.5% through cycles 1–12. Accordingly, the percentage of women who had an absence of scheduled (withdrawal) bleeding through cycles 1–12 averaged 23.5% (Fig. 3). Among women who had any scheduled (withdrawal) bleeding, the mean duration of the withdrawal bleed per cycle ranged from 4.7±4.0 days (cycle 1) to 4.0±2.2 days (cycle 12). The majority of women (60.6%–73.1%) rated the maximum intensity of their scheduled (withdrawal) bleeding as spotting or light. Only a small number of women (0.5% [in cycle 9] to 6.6% [in cycle 2]) rated the maximum intensity of their scheduled (withdrawal) bleeding as heavy.

Proportion of women treated with estradiol valerate/dienogest with an absence of scheduled (withdrawal) bleeding during cycles 1–12 of treatment (full analysis set).
Onset of scheduled (withdrawal) bleeding in cycles 2–12 for women who did bleed occurred in the first 3 days following withdrawal of DNG (i.e., between days 25 and 27 of the treatment cycle) as follows: 12.8% on day 25, 24.9% on day 26, and 15.3% on day 27. The median number of days to onset of scheduled (withdrawal) bleeding through cycles 2–12 was consistently 2.0 (interquartile range 3.0 [1st quarter 1.0, 3rd quarter 4.0]). However, because of a few outliers, the mean number of days from the end of exposure to DNG ranged from a mean 3.8±5.8 days (cycle 6) to 5.4±7.3 days (cycle 3).
Intracyclic bleeding
As noted earlier, because of the protocol instructions on when to start treatment and the definition of intracyclic bleeding, virtually all women (97.1%) experienced intracyclic bleeding during cycle 1. Thereafter, intracyclic bleeding rates diminished over time from 28.8% (cycle 2) to 11.2% (cycle 11) (Fig. 4).

Proportion of women treated with estradiol valerate/dienogest with unscheduled (intracyclic) bleeding during cycles 1–13 of treatment (full analysis set).
The maximum intensity of intracyclic bleeding was most often rated as spotting or light. During treatment cycles 2–13, 40.5% (cycle 3) to 62.2% (cycle 12) of women who experienced intracyclic bleeding rated its intensity as spotting. During cycles 2 to 13, heavy intracyclic bleeding was reported by fluctuating percentages of women (from 0.0% in cycles 5 and 12 to 7.0% in cycle 6).
Safety and tolerability
The most frequently reported AEs in the full analysis set over the entire study, including the extension phase, were headache (15.7%), metrorrhagia (13.1%), and nasopharyngitis (10.4%). Subjects rated most AEs as moderate or mild in intensity.
Just over one-half of subjects (51.8%; 254/490) experienced at least one AE that was considered by the investigator to be at least possibly treatment related (i.e., deemed to be possibly, probably, or definitely related to treatment). The drug-related AEs reported by the greatest number of subjects were metrorrhagia (16.7%); headache, including migraine (8.4%); and amenorrhea (7.3%) (Table 2).
Women are counted only once for each adverse event.
Fifteen serious AEs (SAEs) were reported in 10 subjects; all subjects recovered from their SAEs. Only one SAE (ovarian cyst rupture) was considered by the investigator following FDA criteria to be possibly related to the study medication. None of the other events were deemed to be related to study treatment.
Over the course of the first 13 cycles of treatment, 68 women (13.9%) prematurely discontinued treatment because of AEs. The most frequent AEs leading to discontinuation during the first 13 cycles were metrorrhagia (2.2%), acne (1.6%), and headache (1.2%). Only 5.1% of women in the current study discontinued treatment over the first 13 cycles because of any menstrual-related AEs (i.e., metrorrhagia, menstrual disorder, menorrhagia, dysmenorrhea, dysfunctional uterine bleeding, and amenorrhea). Of the women who entered the extension phase of the study, 5 discontinued study medication because of AEs.
No meaningful changes in participants' vital signs were observed during the study. Mean change at 13 cycles from baseline in body weight was +0.5 kg (standard deviation [SD], 4.3 kg) and at 28 cycles was +0.8 kg (SD, 4.8 kg); only 2 women discontinued due to weight gain.
Over the 13 cycles of treatment, 68% of women reported that their physical well-being fell into one of the following categories: unchanged, somewhat better, or much better than their pretreatment status (22% of women did not respond). Similarly, 68% of women reported that their emotional well-being was unchanged, somewhat better, or much better than their pretreatment status; 22% of women did not respond.
Discussion
This Phase III trial demonstrated that a COC combining E2V and DNG was associated with reliable contraceptive efficacy in women aged 18–35 years in North America. The Pearl Index in this study is consistent with the Pearl Index that was reported in women aged 18–35 years participating in an identically designed, somewhat larger, 20-cycle study conducted in Europe. 15 The Pearl Indices reported in the current study and in the earlier European trial are within the historically reported Pearl Index range for COCs containing low doses of EE. 16
The bleeding profile achieved with this formulation of E2V/DNG appears to be better tolerated than earlier experimental oral contraceptive formulations utilizing E2. Only 5% of women discontinued our study owing to bleeding-related problems over 13 cycles, compared indirectly with data from other studies for discontinuations rates owing to bleeding-related problems of 10%–42% over 12 to 15 cycles with the earlier E2 formulations. 4,5,17 An earlier study with an E2-based formulation, which was subsequently approved in Finland in women aged >40 years, 18 reported a discontinuation rate owing to bleeding problems of 9% over 13 cycles. 19 The low discontinuation rate owing to bleeding problems found in the current trial was also very similar to discontinuation rates at 1 year owing to bleeding problems for women using a COC containing E2 and nomegestrol acetate (NOMAC), which was reported to be 4% in two studies. 20,21 One-year discontinuation rates owing to bleeding problems can range up to 9% for some approved low-dose EE-based formulations. 22,23
Intracyclic bleeding was reported in approximately 11% to 29% of women participating in the current trial (excluding data from cycle 1). When it occurred, the intensity of intracyclic bleeding was most often rated as spotting. This is similar to the results from an earlier seven-cycle European study, which compared E2V/DNG with a COC containing EE 20 mcg/levonorgestrel (LNG) 100 mcg in 798 women. 24 In that study, the rate of intracyclic bleeding with E2V/DNG (10.5%–18.6%) was comparable to that observed with EE/LNG (9.9%–17.1%). In addition, the occurrence of scheduled (withdrawal) bleeding was lower with E2V/DNG than with EE/LNG, 24 across cycles 1–7; an absence of scheduled (withdrawal) bleeding was reported in an average of 19.4% and 7.7% of E2V/DNG and EE/LNG recipients per cycle, respectively. The proportion of women with absent scheduled (withdrawal) bleeding in cycles 1–12 in the current study averaged 23.5% and appears somewhat higher (through indirect comparisons across studies) compared with the findings of the comparative European study (19.4% across cycles 1–7). In both studies, the intensity of scheduled (withdrawal) bleeding was frequently considered to be spotting or light. Given that E2V/DNG has a slightly higher rate of absent scheduled (withdrawal) bleeding than a COC containing EE/LNG, 24 women considering the use of this formulation should be counseled appropriately. Currently, E2V/DNG is the only COC with an FDA-approved indication for treatment of heavy menstrual bleeding in women who choose to use an oral contraceptive for birth control.
The new dosing used in this formulation to achieve good cycle control requires different patient counseling. Women are to start the first pill on the first day of menses. This formulation results in higher than traditional rates of no bleeding or spotting and withdrawal bleeding that is perceived to be light. In addition, the onset of the bleeding generally precedes the placebo pills but may be delayed until after the woman starts her next pack of pills. Each of the differences needs to be explained to the patient in advance so that she will not be unduly concerned by these changes in her bleeding patterns.
The current study's limitations need to be considered when making comparisons to other studies or translating the relevance of the outcomes to routine clinical practice. Because it was a noncomparative study, only indirect comparisons can be made to other studies. As with all clinical trials, subjects were carefully screened based on the strict inclusion/exclusion criteria, which may not necessarily reflect typical users in practice. Compliance to treatment during the study may be higher than that typically observed in routine practice, owing to regular monitoring or increased motivation through regular contact with study staff, which may in turn affect efficacy and continuation rates. Finally, the study recruited only a limited number of women, which does not allow for a robust evaluation of the frequency of rare and very rare events. An FDA-directed, large-scale post-marketing epidemiological study has been initiated (ClinicalTrials.gov Identifier: NCT01009684) to evaluate both the short- and long-term safety of E2V/DNG in typical use.
Conclusion
E2V/DNG provides reliable contraceptive efficacy and reasonable cycle control in healthy, non-obese women aged 18–35 years in North America. E2V/DNG offers another alternative for women who choose a COC as their method of birth control.
Footnotes
Acknowledgments
This study was funded by Bayer HealthCare, Berlin, Germany, the manufacturer of Natazia®/Qlaira®. The authors would like to thank Lyndal Staples of inScience Communications, Springer Healthcare, for undertaking a technical edit of the manuscript. This work was funded by Bayer HealthCare, Inc., Wayne, New Jersey.
Author Disclosure Statement
Anita Nelson has participated in speakers' bureaus and has worked as a consultant on advisory boards for Bayer HealthCare, and her clinic has received research grants from that company. Susanne Parke, Uwe Mellinger, Edio Zampaglione, and Anja Schmidt are employees of Bayer HealthCare.