
Abstract
Abstract
Surgical innovation involves the conceptualization, research, and translation of a novel idea into a viable procedure or device. The technological advancements made within the field of pediatric surgery over the last century have led to major improvements in patient care and outcomes. There has, however, been a parallel increase in the complexity of the regulatory bodies governing research and device implementation. This article briefly outlines the history of innovation in pediatric surgery, describes the existing regulatory bodies governing surgical research and device development (i.e., Department of Health and Human Services, Food and Drug Administration), and offers a set of guidelines for the pediatric surgeon planning to incorporate a new procedure or device into clinical practice.
Prologue
Introduction
Surgical innovation involves a combination of the conceptualization, research, and translation of a novel idea into a viable procedure or device. Successful surgical innovations such as the Fogarty catheter, cardiopulmonary bypass, or extracorporeal membrane oxygenation improve or reinvent the existing approach to a clinical problem and ultimately improve outcomes.
The 19th century brought two significant transformational innovations in the field of surgery, the advent of ether anesthesia credited to William Morton and the development of antiseptic surgery pioneered by Joseph Lister.1,2 Many congenital malformations that were previously inoperable became curable as novel surgical procedures were introduced. Formal risk assessment, animal models, or professional workshops were typically nonexistent as the alternative to swift and heroic operative intervention was death or permanent disability. The need to address safety and outcomes was at best an afterthought. Innovations in the 20th century, however, were largely incremental improvements on established procedures for which safety and outcomes were already established. The potential to compromise these (sometimes unsubstantiated) “gold standards” of efficacy and safety heralded the need for more rigorous paths to innovation. Surgical innovators of the 21st century have further increased the complexity of procedures by introducing advanced technologies such as minimal access surgical devices, raising concerns over the safety of implementing such tools. With the adoption of advanced technologies has come a paralleled increase in complexity of the regulation of novel procedures.
Surgeons, perhaps more so than any other medical specialists, should carefully balance the risks inherent to their devices and/or procedures with their potential benefits. For pediatric surgeons, defining this balance becomes even more difficult due to the invasive nature of the procedures being performed on very young and often very sick children. As innovation in pediatric surgery continues to push the technological envelope, it is important that members of our community adhere to the highest moral standards. This article discusses the current state of the regulatory processes governing devices and research in children and provides a set of recommended guidelines for implementing new technologies into one's surgical practice.
Existing Regulatory Bodies
In the United States, pediatric research falls under the regulation of institutional review boards (IRB) and the United States Department of Health and Human Services (DHHS), which serve the purpose of upholding the guidelines set forth by state and federal legislative bodies. The Food and Drug Administration (FDA) regulates the use of all surgical devices within the United States. Although the majority of pediatric surgeons will not design large clinical trials or novel devices, it is helpful to understand the regulatory processes when implementing new techniques or devices into practice. Of note, other countries have a multitude of local and national regulatory bodies, but for simplicity we will focus on the United States model.
DHHS pediatric research guidelines
The DHHS categorizes pediatric research based on both the degree of risk and the potential benefit to the study participants. 3 The DHHS outlines four successive categories of pediatric research labeled as codes 46.404–46.407 (Table 1). The first three of these codes encompass studies with potential for benefit to the participant with relatively low levels of risk exposure. The fourth DHHS code, 46.407, includes research studies that expose the participants to potential risk in the absence of direct or indirect benefit, but have the potential to benefit children in general.
Individual IRBs oversee studies that fall under the codes 46.404–46.406. A study that falls under the category 46.407, however, may not be approved solely by an IRB. The Secretary of the DHHS reviews all proposals that fall into this category. The Secretary decides, after consulting with experts in the proposed field of study, whether or not to approve the study in question. 4
Importantly, all pediatric research proposals regardless of which DHHS code they fall under should demonstrate an appropriate process for obtaining both patient assent and parent/guardian consent as defined by The American Academy of Pediatrics Committee on Bioethics. 5 The currently accepted standard of care is to obtain patient assent before enrollment in a study when feasible (i.e., when the patient is developmentally capable of affirming participation after receiving a cognitive age-appropriate explanation of the study/procedure, risks, benefits, and alternative options). Parental permission/consent is required whenever possible (i.e., nonemergent settings) if the patient is a nonemancipated minor. Practically speaking, parental permission/consent involves all of the components of informed consent in an adult population.
The dynamic continuum of early cognitive development implies that the requirement for obtaining pediatric patient assent should be assessed on a case-by-case basis. When the proposed intervention or clinical trial has the potential for direct or indirect benefit (i.e., DHHS codes 46.404–46.406), parental permission/consent may be weighed more heavily than patient assent. Patient dissent, however, is generally considered more significant in procedures or trials that lack the potential for patient benefit (i.e., DHHS code 46.407). 6 Given the complexity of these situations, discussion with members of the IRB or other similar regulatory bodies may be considered to ensure that a study protocol is in compliance with the applicable regulations.
FDA device approval
The FDA categorizes new devices into three classes based on the potential risk incurred by using the device in humans. Class I devices pose minimal harm to the recipient and do not typically require premarket notification or approval (i.e., clinical data supporting safety and efficacy). Many surgical staplers and endoscopic instruments are Class I devices. Class II devices pose an intermediate level of potential harm to the recipient and should meet a set of defined performance standards before being approved for general use. This typically implies showing clinical efficacy comparable to similar existing devices. Many surgical prostheses that do not support vital functions, such as mesh implants, are Class II devices. Class III devices pose significant potential harm to the recipient and require premarket approval with clinical data supporting both safety and efficacy. Mechanical prostheses such as heart valves and orthopedic implants are Class III devices. 7
The collection of preliminary data for nonFDA approved devices is regulated by the IRB. If the IRB determines that the device provides insignificant risk to the study participants, the study may proceed. However, if the IRB concludes that the proposed study exposes the participants to significant risk, the FDA should approve an investigational device exemption before commencement of the study. 8
Importantly, for pediatric devices that fall into the Class III category and require premarket approval, there is a means to bypass the long and costly process of clinical trials. Devices intended to treat conditions that affect less than 4000 people per year in the United States qualify for humanitarian device exemptions (HDE). This allows approval of such devices when safety has been demonstrated, and the probable benefits outweigh the risks of using the device. 7,8 The HDE prevents delays in disseminating technologies due to the inability to properly power premarket clinical trials. The HDE does not, however, decrease the importance of performing the necessary clinical trials after FDA approval.
To summarize, the implication for the pediatric surgeon is that before using a non-FDA approved device in humans, one should obtain IRB approval. If the IRB believes that there is sufficient patient risk associated with the use of the novel device, the investigator should apply to the FDA directly for an investigational device exemption. After initial clinical data are obtained, the investigator may apply for FDA approval. In the case of devices intended for use in rare pediatric conditions, the investigator may apply to the FDA for an HDE to more rapidly disseminate the technology.
Proposed Guidelines for Innovation in Pediatric Surgery
The DHHS and FDA provide detailed regulations regarding the approval of clinical research and medical device use as just described. They do not, however, guide the pediatric surgeon in the incorporation of new techniques or devices into daily practice. The following paragraphs outline a set of guidelines to aid in that process. These guidelines are not meant to be prescriptive, but rather to act as a set of principles that surgeons may consider when contemplating a new procedure or use of a new device.
The initial branch point in the algorithm (Fig. 1) distinguishes the implementation of an existing procedure or device from an entirely new procedure or device. Consider the implementation of single incision laparoscopic surgery (a novel technology already performed at many institutions) versus the introduction of an entirely novel procedure/device such as a hypothetical device for achieving a transoral esophageal anastomosis for esophageal atresia. In the first scenario, it is recommended that an experienced surgeon participate in a mentorship or observational experience before proceeding, whereas a less experienced surgeon may wish to enroll in a formal technical workshop. After an appropriate level of technical competence (as determined by the individual surgeon and/or her institutional peers) is achieved, one may consider making their intentions known to their departmental chair or equivalent through a formal letter of intent before performing the procedure independently.
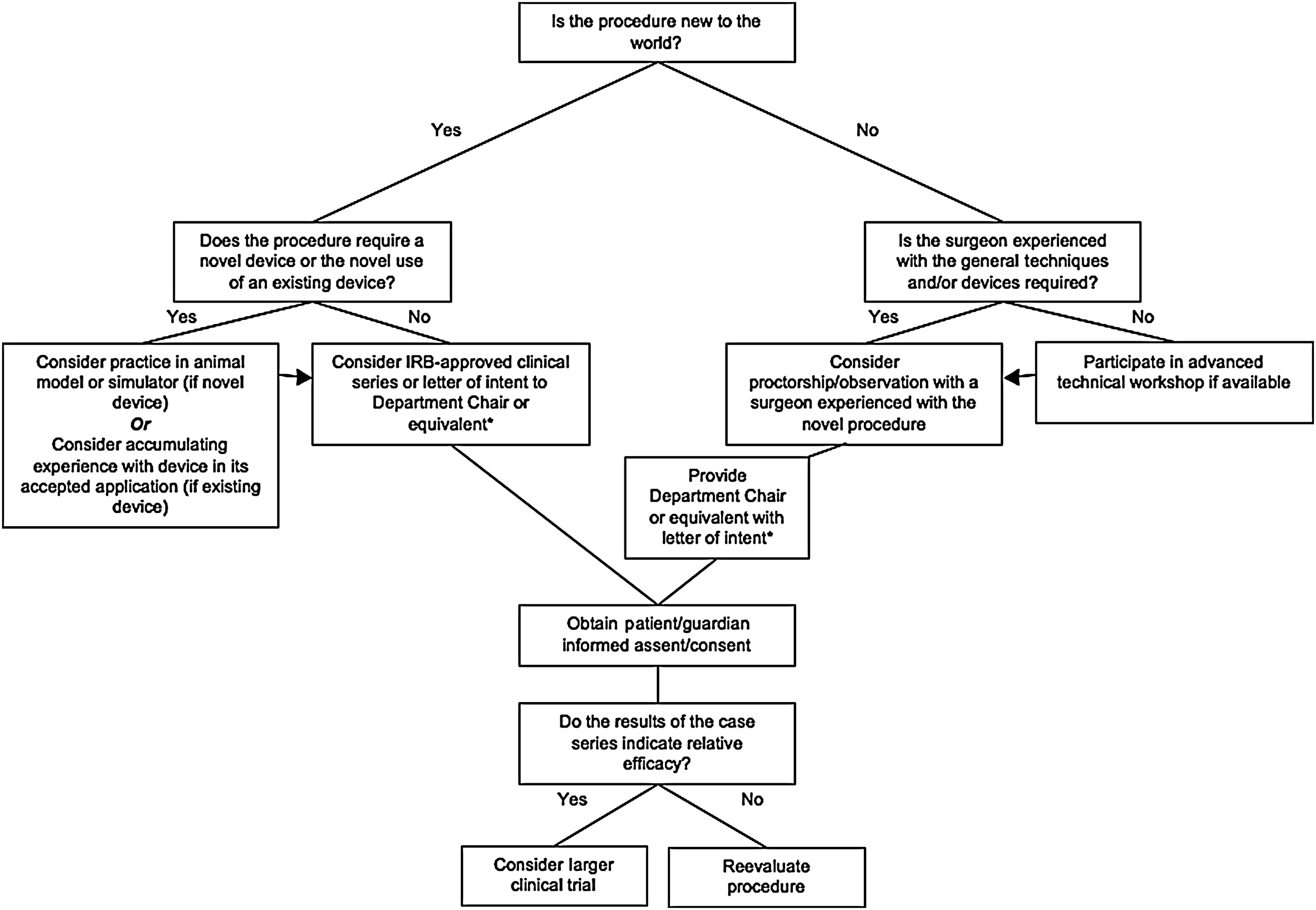
Guidelines for implementation of a new procedure or device. IRB, institutional review boards. *Such a letter would include a description of existing foundational technique(s), a description of the novel technique, a summary of the published results comparing the novel technique to existing technique(s) if available, and a description of the preparation undertaken by the surgeon prior to attempting the novel procedure.
When a procedure or device in question is new to the world, as in the case where a transoral esophageal anastomosis is created with a novel device, the recommended pathway is slightly more involved. The first branch point of the algorithm in this situation (the left side of Fig. 1) is navigated based on whether the new procedure requires the use of a novel device. A subcategory exists when the procedure involves the use of an existing device in a new way.
First, when no device is required, it is recommended that before attempting the procedure in a patient the surgeon considers obtaining IRB approval as a means of verifying his or her assessment of potential clinical utility with an unbiased third party. The necessity of IRB approval in this case depends on the complexity of the proposed procedure and the potential risk/benefit ratio. For low risk procedures, it may be reasonable to skip this step.
The next situation is that of a novel procedure requiring the use of an existing device in a new way. A past example relevant to pediatric surgery is the ArthoKnife (ConMed Linvatec and Hall® Surgical), a device intended and approved for use in arthoscopic orthopedic procedures that was, until recently, used for the laparoscopic pyloromyotomy (the device is no longer manufactured). In this situation, FDA approval had already been given for the device in question, but not for this particular application. This is considered “off-label” use. It is then the surgeon's prerogative to attain technical competency with the device before using it in an innovative fashion. The next step would then be to consider IRB approval and/or discussion with one's departmental superiors before proceeding clinically with the novel application. If the new application deviates only marginally from the approved use, this last step may be unnecessary. Interestingly, it is unlikely that most surgeons who adopted this device for pyloromyotomy went through any formal process to use it.
The third and most complex situation is with the use of an entirely novel surgical device. Initial use of the device in an animal model should be considered to provide the surgeon with hands-on training before use in humans. Further, the resultant animal data would support any subsequent IRB or FDA proposals. It is important to keep in mind that the purpose of a preclinical and/or a premarket trial is to prove safety and efficacy, described as the ability to achieve results comparable to existing technology.
The subsequent steps leading to the application of a novel device or procedure are intuitive and are undoubtedly familiar to the practicing pediatric surgeon-innovator. After technical proficiency has been achieved and Chairperson, IRB, and/or FDA approval has been given, one should identify the appropriate surgical candidate and obtain both patient assent and parental consent as just described (see section on DHHS pediatric research guidelines ). If an initial case series indicates efficacy either by improving on clinical outcomes or by providing comparable outcomes (to the status quo) with an associated financial or procedural benefit, it becomes important to engage in sufficiently powered clinical trials.
Institutional Implementation of Innovation Guidelines
The guidelines just described are intended to form the basis of more in-depth conversations among surgeons at individual institutions, so that they can develop specific policies that are best suited to their environment. The goal is to maintain patient safety and address regulatory issues, without stifling the creativity and autonomy of the individual practitioners. There is no doubt that achieving this balance is a unique challenge for the leadership of any organization.
Having a program dedicated to pediatric medical device development, as well as a thematic emphasis on innovation in minimal access surgery, has necessitated innovation guidelines at our institution. For example, the Multidisciplinary Initiative for Surgical Technology Research, a joint venture between Stanford University, Lucile Packard Children's Hospital (LPCH) and SRI International (an engineering research institute), uses the proposed guidelines for device development. Multidisciplinary Initiative for Surgical Technology Research carefully assesses the safety implications of novel device concepts based on input from surgeons at LPCH and elsewhere, as well as by comparison to existing devices with similar capabilities. When a prototype is ready for testing, we then work closely with the FDA and the local IRB to determine the appropriate path to clinical testing.
Guidelines for introducing innovative procedures are also utilized at LPCH. For example, when laparoscopic partial splenectomy was first performed by the senior author at LPCH, the procedure was new to the institution. Open partial splenectomy was described in the literature, but the use of laparoscopy for this purpose was not. The operating surgeon, however, did have previous experience with the laparoscopic approach at another institution where it was first performed. Therefore, the surgeon prepared a document that outlined the procedure, summarized the existing literature, and described the underlying technical experience. This document was then reviewed by the department chair and discussed among surgical peers so that any concerns could be addressed before proceeding with the procedure. There was also a full discussion around informed consent with the patients. Of note, if the laparoscopic approach had not ever been previously performed, the surgeon could document previous underlying component expertise in advanced laparoscopy and in splenectomy, forming the basis of accountability for the innovation of combining the two skills.
Summary
As pediatric surgery has evolved, many of the innovative techniques and devices being researched now serve the purpose of more efficiently treating certain diseases and malformations as opposed to simply providing an alternative to death. This distinction carries implications for the process of surgical innovation and the importance of balancing the risks of clinical research with the potential benefits of new interventions. The recommended guidelines just introduced are meant as an aid to the pediatric surgeon when navigating through the rewarding process of surgical innovation.
Footnotes
Disclosure Statement
No competing financial interests exist.