
Abstract
Abstract
The protein mammalian target of rapamycin (mTOR) plays a central role in cell growth and proliferation. Excessive mTOR activity is a prominent feature of many neoplasms and hamartoma syndromes, including lymphangioleiomyomatosis (LAM), a destructive lung disease that causes progressive respiratory failure in women. Although pharmacological inhibitors of mTOR should directly target the pathogenesis of these disorders, their clinical efficacy has been suboptimal. Recent scientific findings reviewed here have greatly improved our understanding of mTOR signaling mechanisms, provided new insights into the control of cell growth and proliferation, and facilitated the development of new therapeutic approaches in LAM, as well as other neoplastic disorders that exhibit excessive mTOR activity.
Introduction
The Molecular Organization of mTOR
Initially reported in 1994,8–10 mTOR is a ∼240-kDa protein composed of structural domains that permit multiple protein–protein interactions, and the assembly of macromolecular signaling complexes that control anabolic functions 11 (Fig. 1). A C-terminal PI3K-like domain contains the serine/threonine kinase active site. The heterogeneity of its target sequences suggests that the mTOR kinase domain directly phosphorylates certain effector substrates, but may control the phosphorylation of others via intermediary kinases or phosphatases. 12 Of the few known post-translational modification sites in mTOR, all involve serine/threonine phosphorylation.13,14 The auto-phosphorylation of serine 2481 (S·2481) correlates with mTOR kinase activity, but not necessarily the ability to activate downstream effectors. 15 Although it inhibits mTOR signaling, rapamycin does not necessarily reduce mTOR autokinase activity. Novel mTOR inhibitors are currently undergoing characterization.16–18 The phosphorylation of S·2448 also correlates with mTOR kinase activity, as well as with mitogen or nutrient levels.19,20 Phosphorylation of S·1261 in cells exposed to insulin is required for the activation of mTOR, its autophosphorylation at S·2481, and its regulation of cell size. 21
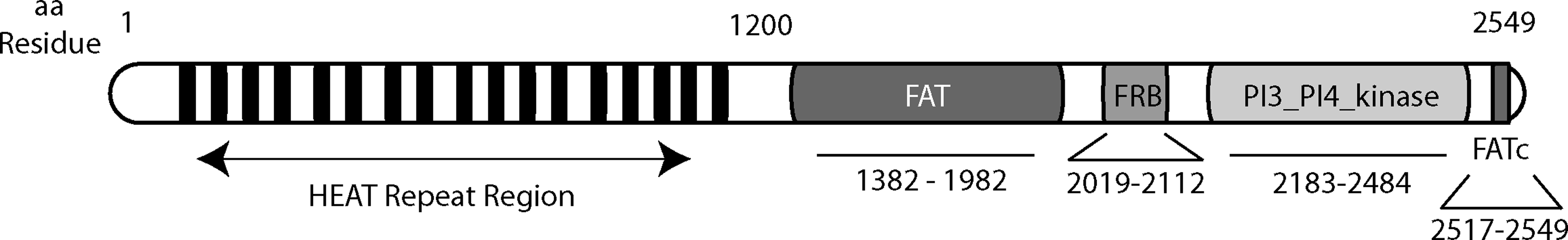
Structural domains and motifs in mTOR. mTOR consists of 2549 amino acids organized in several structural domains. The N-terminal portion of mTOR contains multiple HEAT (
mTOR also contains amino acid residues that mediate its subcellular localization. mTOR nuclear trafficking requires two leucine residues in its HEAT repeat region (Fig. 1); constitutive nuclear localization of mTOR is observed in several tumor cell lines.22,23 A distinct set of residues in the HEAT repeat region determines mTOR localization to endoplasmic reticulum (ER) and/or Golgi membranes. 24 The ER may provide a physical platform for mTOR metabolic sensing functions, control of the ER stress response, 25 and coordination of mTOR signaling to the translational and transcriptional machinery.26,27 mTOR can also localize to mitochondrial membranes, although the molecular mechanism is unknown. 28 mTOR mitochondrial localization was associated with an effect on ATP production and oxygen consumption. 29 Furthermore, mTOR directly sensed changes in cell redox state, possibly via highly-conserved cysteine residues in its FATc domain.30,31
The Core mTOR Complexes
From yeast to mammals, TOR nucleates two distinct macromolecular complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2)32,33 (Fig. 2). mTORC1 is defined by the presence of ‘regulatory associated partner of mTOR’ (raptor). Raptor physically links mTOR to its effectors (e.g., 4E-BP1, p70 S6 kinase (S6K)) by binding their conserved ‘target of rapamycin signaling’ (TOS) motifs.34–36 mTORC1 enhances protein synthesis and ribosomal biogenesis, whereas it suppresses catabolic responses such as autophagy and the induction of cell cycle inhibitor genes.
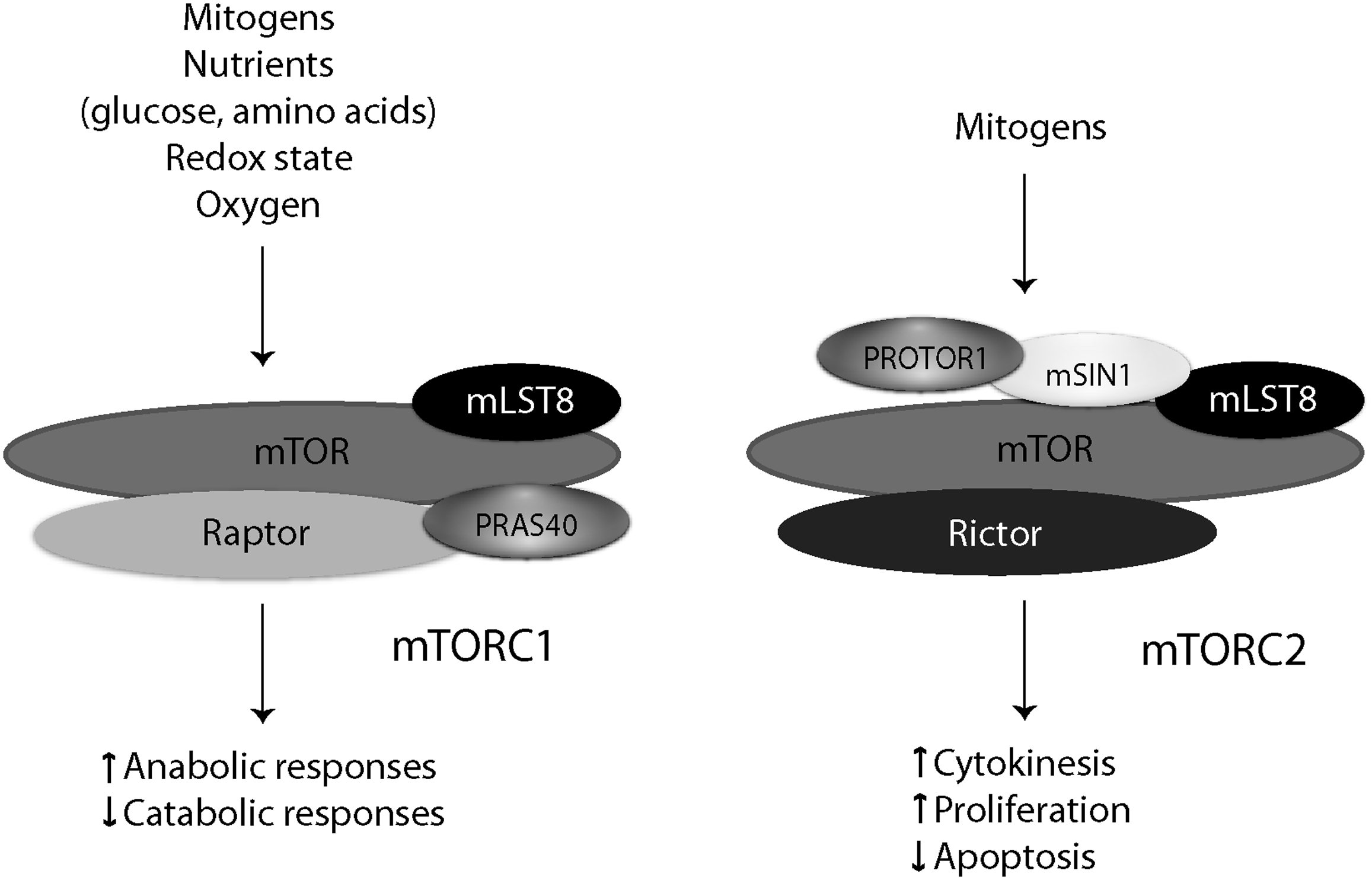
Composition of the mTOR core complexes. mTOR complexes integrate mitogenic and metabolic signals, and control anabolic or catabolic cellular functions as appropriate. mTORC1, as defined by the presence of raptor, is shown to the left of mTORC2, which is defined by its association with rictor. Other adapter proteins that coordinate the functions of mTOR complexes are also shown.
mTORC2 is defined by the presence of ‘rapamycin insensitive companion of mTOR’ (rictor), and regulates cell proliferation, polarity, and cytokinesis by phosphorylating the hydrophobic and/or turn motifs on a subset of ‘cAMP-dependent protein kinase/protein kinase G/protein kinase C’ (AGC kinases; e.g., protein kinase Cα, Akt) kinases. 37 Other known mTORC2-specific proteins include mammalian SAPK-interacting protein 1 (mSIN1), and ‘protein observed with rictor-1’ (PROTOR1). Rictor and mSIN1 determine mTORC2 substrate specificity, and are required for the phosphorylation of Akt or PKCα.38–41 PROTOR1, also known as PRR5, appears to regulate platelet-derived growth factor (PDGF) signaling and cellular apoptosis.42–44 Both mTOR complexes contain mLST8 (originally termed GβL), which is essential for the function of mTORC2, and modulates mTORC1 activity in nutrient-sensitive manner. 45
Regulated mTORC–Protein Interactions
The small GTPase Rheb is an essential binding partner and activator of mTOR. GTP-bound Rheb stimulates mTORC1 activity. The GDP-bound (inactive) state is favored by the C-terminal GAP activity of tuberin (TSC2), but does not appear to affect the interaction between Rheb and mTOR.46–49 TSC2 is therefore an endogenous suppressor of mTORC1. In contrast, TSC2 appears to positively regulate mTORC2 independent of its GAP activity or Rheb.50,51
Additional mTOR binding partners act as ‘signal input receivers' for the mTOR complexes. The mTORC1 protein ‘proline-rich Akt/PKB Substrate 40’ (PRAS40) contains a TOS (i.e., raptor-binding) motif, and appears to be a bone fide mTORC1 substrate.52,53 However, PRAS40 also constitutively binds and inhibits mTORC1, dissociating from the complex in cells exposed to insulin or nutrients.43,54,55 The integral ER membrane protein FKBP38 constitutively inhibits mTORC1 activity by blocking the interaction between mTOR and Rheb.56–58 FKBP38 dissociates from Rheb in cells exposed to mitogens, and thus facilitates the activation of mTOR. Phospholipase D (PLD), a mitogen-activated Rheb- and mTOR-binding protein, catalyzes the synthesis of phosphatidic acid, which directly activates mTOR via binding to the FRB domain.59–61 The rapamycin/FKBP12 complex might inhibits mTOR in part by competing for the phosphatidic acid-binding site in the FRB domain. 62 Ras-related GTP binding (Rag) GTPases were also recently identified as mTOR-associated proteins. 63 Rag heterodimers targeted mTOR to cytoplasmic vesicular structures in response to amino acid repletion; their possible effects on mitogen-dependent mTOR signaling have not been defined.
mTOR interacts with, and regulates, the protein phosphatase 2A catalytic subunit (PP2Ac) via its associated adaptor subunit α4. In yeast, TORC1 controls the nuclear localization of transcription factors by associating with, and likely phosphorylating, the α4 homologue Tap42.64,65 α4, or Tap42, can be negative64,66,67 or positive68–70 regulators of PP2Ac, which in turn can dephosphorylate multiple targets, including AGC kinases. 71 Some propose that mitogen-stimulated phosphorylation of S6K at its T·389 residue results from the inhibition of PP2Ac activity by mTOR. 66 Genetic studies indicate a role for α4 in certain functions attributed to mTORC2, such as cytokinesis and cell spreading.72,73
Signaling Inputs to the mTOR Complexes
Mitogen-activated and metabolic sensing pathways converge upon the core mTOR signaling components at multiple levels (Fig. 3). The best studied is that involving PI3K, phosphatidylinositol-dependent kinase-1 (PDK1), and Akt. Ligation of growth factor receptors permits the recruitment and activation of IRS-1 and PI3K. Phosphorylated phosphatidylinositols (PI3Ps) bind PDK1, which then phosphorylates Akt in its activation loop (T·308). Akt phosphorylates TSC2, suppresses its GAP activity, and relieves the inhibitory effect of TSC2 on Rheb and mTORC1 activity.74,75 Commonly mutated in neoplastic disorders, the protein phosphatase and tensin homologue (PTEN) is a PI3P phosphatase; its loss results in elevated PDK1, Akt, and mTOR activity. PDK1 can also directly phosphorylate the activation loop of other AGC kinases that undergo phosphorylation by mTOR at their hydrophobic motifs (e.g., PKCα, PKCδ, S6K).38,39,76–78 Therefore, the PI3K/PDK1 pathway can activate AGC kinases independently of TSC2 or Rheb. The concurrent phosphorylation of AGC kinases by PDK1 and mTOR may be required for their maximal activation or stability. 37 In agreement, the ability of rapamycin to block tumor progression or apoptosis in mice varied according to PI3P levels or Akt activity, suggesting that the net cellular growth and proliferation of neoplastic cells depends on the relative activities of the PI3K and mTOR signaling pathways.79–82

Coordination of mTORC1 and mTORC2 signaling activities. mTOR signaling can be organized into concentric spheres consisting from center to periphery of the mTORCs (center), their regulatory interaction partners, proteins that indirectly modify mTORC activity or mediate mTORC functions, and the mTORC-related physiological stimuli or functions. Proteins that modify mTORC activity are above the horizontal line, whereas those which mediate mTOR functions are below. Although multiple feedback and crosstalk mechanisms occur, the directionality of mTOR signaling is depicted in the center, with the mTOR complexes acting as signal integrators. Color images available online at www.liebertonline.com/lrb.
Whereas Akt enhances mTORC1 activity, its S·473 residue is also a substrate for mTORC2. 83 The biological significance of Akt as both a substrate of mTORC2 and a kinase for mTOR signaling intermediates remains unclear. Akt appears to act as a signal transducer of growth factor signaling in the control of mTORC1 and cell growth, while simultaneously mediating mTORC2 suppression of apoptosis and enhancement of proliferation. Future studies investigating the intracellular regulators of mTORC2, as well as the subcellular localization and phosphorylation kinetics of the respective mTOR complexes may shed light on their relative contributions to the balance between cell proliferation and apoptosis.
mTORC1 simultaneously controls a negative feedback mechanism that dampens growth factor signaling. The mTORC1 effector S6K phosphorylates and inhibits IRS-1, thereby limiting the activation of mTOR in TSC2-deficient tumors84,85 (Fig. 3). This may in part explain the relatively benign nature of TSC and LAM hamartomas, as well as the limited clinical efficacy of rapamycin. 86 That is, by inhibiting mTORC1, rapamycin relieves the S6K-dependent inhibition of IRS-1/PI3K/Akt signaling, and limits its therapeutic effect on tumor growth.
In addition to the PI3K pathway, receptor tyrosine kinases activate Ras, an oncogenic protein that promotes the transformation and growth of neoplasms. Ras activates intermediates, including p90 ribosomal S6 kinase (Rsk), the mitogen-activated protein kinase Erk, and phospholipase D, all of which can modulate mTOR signaling (Fig. 3). TSC2 and raptor contain Erk- or Rsk-phosphorylated residues.87–90 Like its negative feedback effect on the PI3K/Akt pathway, S6K also attenuated Erk activity, and Erk was ‘de-repressed’ in cells exposed to rapamycin. 91
Other inputs to the TSC2/Rheb/mTOR signaling axis include metabolic sensing pathways, which are commonly dysregulated in cancer or hamartoma syndromes.6,92 The energy (ATP)-sensing protein AMP-dependent protein kinase (AMPK) can control TSC2 or raptor via direct phosphorylation.93–95 Its upstream kinase LKB1 is lost or mutated in a variety of cancers. 5 Phosphorylation of TSC2 by AMPK permits its phosphorylation by GSK3, an intermediate in the Wnt signaling pathway.94,96 Thus, Wnt, an important controller of differentiation and proliferation, can activate mTORC1, cell growth, and tumor progression independent of its known effect on the β-catenin-mediated Wnt transcriptional program. 97
mTORC Signaling Targets in LAM and Neoplasia
Translation and cell growth
Excessive cell growth and proliferation require a robust protein synthesis machinery.98,99 In response to mitogens, S6K and 4E-BP1 associate with mTORC1 via their respective TOS motifs. Phosphorylation of 4E-BP1 at multiple sites inhibits its interaction with the mRNA CAP-binding protein eIF4E, and permits the initiation of translation. 100 This mechanism recognizes a subset of mRNAs, many of which are involved in cell growth, proliferation, and angiogenesis (e.g., ribosomal proteins, cyclin D1, HIF-1α). eIF4E was sufficient to transform cells, 101 and overexpression of 4E-BP1 partially reversed v-src-induced transformation. 102 S6K phosphorylates components of the translational machinery, and promotes ribosomal biogenesis via the transcriptional induction of ribosomal RNA and genes encoding ribosomal proteins.
Apart from S6K and Akt, mTORCs can associate with and phosphorylate two other AGC kinases that modify cell proliferation and apoptosis: serum/glucocorticoid kinase-1(SGK1) and protein kinase Cδ.78,103,104 Genetic or biochemical studies in C. elegans and mammalian cell lines indicate phosphorylation and regulation of SGK by TORC2 and not mTORC1.105–107 In a cell line overexpressing mTOR, however, SGK was phosphorylated by mTORC1, and subsequently blocked cell cycle arrest. 108 In contrast to the mTORC2 substrate PKCα, PKCδ is phosphorylated by mTORC1, phosphorylates 4E-BP1, and controls the initiation of translation. 104 Interestingly, PKCδ is a kinase for the transcription factor STAT1 kinase, and was detected in a rapamycin-sensitive macromolecular complex with mTOR and STAT1.109–111
Gene transcription
Neoplasms exhibit transcriptomic programs that determine oncogenic potential and pathogenesis.112,113 Emerging data indicate that mammalian or yeast TOR can localize to the nucleus, bind DNA regulatory elements, and modify the transcription of nutrient and stress response genes.114–116 mTOR can physically associate with transcription factors that control cell proliferation, apoptosis, ribosomal biogenesis, and angiogenesis. In response to growth factors, mTORC1 can associate with and phosphorylate STAT3, an oncogenic transcription factor that is increased in LAM, and a variety of cancers.117–119 Conversely, inactivation of mTOR or its associated phosphatase complex, promoted its association with STAT1, enhanced STAT1 nuclear content, and amplified the transcription of STAT1-dependent anti-proliferative and pro-apoptotic genes. 109 The balance between STAT1 and STAT3 is dysregulated in LAM, and is thought to control the fate of neoplastic cells. 120 mTOR also activates NF-κB, a pro-proliferative transcription factor implicated in the pathogenesis of cancer. 121 Others demonstrated a physical association between mTOR and IKKα, the IκB kinase required for NF-κB translocation to the nucleus. 122 Loss of TSC2 inhibited NF-κB-dependent responses to apoptosis inducting agents. 123
The metabolic switch from oxidative phosphorylation to glycolysis is a prominent feature of tumor biology.
92
mTOR physically bound the promoters of mitochondrial biogenesis genes, and acted as a transcriptional co-activator via physical interactions with the transcription factors PGC-1α and YY1.
124
mTORC1 also interacted with hypoxia inducible factor-1α (HIF-1α), a transcription factor that, in cells exposed to hypoxia, controls a gene transcription program of angiogenesis, proliferation, and metabolic switch to anaerobic metabolism.
125
A TOS motif in HIF-1α mediated its interaction with raptor and enhanced transcriptional activity.
126
mTORC1 also promoted the synthesis of HIF-1α protein.
127
Moreover, in a negative feedback mechanism, the hypoxia-induced protein ‘
Other emerging mTOR targets in LAM
Recent studies revealed an important role for autophagy in oncogenesis and tumor survival. 129 mTORC1 interacts with, phosphorylates, and inhibits the activity of Ulk1, a protein that initiates autophagy.130,131 Whereas mTOR inhibitors block cell growth and proliferation, they may limit their own efficacy by promoting autophagy and reducing apoptosis in established tumors.
TSC2-deficient cells clearly exhibit abnormalities in cytoskeletal function consistent with their ability to invade and metastasize.132,133 mTORC2 directly interacted with and regulated Prex1, a Rac1 guanine nucleotide exchange factor that controls cytoskeletal rearrangement. 134
Although this discussion has been restricted to direct mTOR signaling interactions (Fig. 3), certain indirect effects of mTOR on biological processes relevant to LAM are worthy of mention. TSC2-deficient cells exhibit an activated ER stress responses, consistent with their requirement for protein synthesis and cell growth. 25 The ER stress-activated transcription factor ATF6 rendered dormant cancer cells resistant to rapamycin-induced apoptosis and tumor regression. 135 ER stress-activated proteins (e.g., PERK, DAPK, eIF2α, ATF6) may represent potential targets for the sensitization of LAM and neoplastic cells to apoptotic stimuli.
Consistent with the restriction of LAM to women, estrogen promotes the proliferation and metastasis of TSC2-deficient tumors in vivo, or TSC2-deficient cells derived from renal angiomyolipomas, in mTOR-dependent fashion.136–139 Separate studies suggested that prolactin stimulates mTOR, and modifies STAT1 activity in mTOR-dependent fashion.140,141 Further studies are required to better understand how mTORCs interact with androgen or estrogen signaling pathways.
Although the cell of origin in LAM is unknown, the expression of both neuroendocrine and smooth muscle cell lineage markers in LAM cells suggests abnormalities in cellular differentiation. In TSC1-deficient mice, mTOR controlled hematopoietic stem cell quiescence, renewal, and lineage development. Further studies investigating the role of mTOR in the biology of LAM or cancer stem cells will permit a better understanding on the origins of LAM.
Conclusions
Since the identification of the core mitogen-activated mTOR pathway (i.e., PI3K/Akt/TSC2/mTOR), a large number of additional mTOR-interacting proteins and signaling pathways have been identified. It has become clear that neoplastic cells express oncogenes, or lack tumor suppressors, that can render them resistant to the antiproliferative effects of rapamycin. Moreover, the normal feedback mechanisms that limit cell proliferation can be blocked by rapamycin. Finally, TSC2 and Rheb participate in signaling mechanisms other than the mTOR pathway that contribute to the cytoskeletal, 132 hormonal, 137 and proliferative 142 characteristics of neoplastic cells. Further characterization of the mTOR complexes and their signal integration functions will lead to therapeutic approaches for LAM and neoplasia that simultaneously target multiple receptors and signaling intermediates.
Footnotes
Acknowledgments
The author thanks Dr. Joel Moss and Dr. Stewart Levine (National Institutes of Health) for helpful comments and suggestions regarding the content of this manuscript. The author's work is supported by the National Institutes of Health (R01-CA125436) in conjunction with the Tuberous Sclerosis Alliance and Tuberous Sclerosis Canada, an ATS/LAM Foundation Award, and the Canadian Institutes of Health Research.
Disclosures
Dr. Kristof has no conflicts of interest or financial ties to disclose.