
Abstract
Abstract
Research interest in lymphangioleiomyomatosis (LAM) has grown dramatically in the past decade, particularly among cancer biologists. There are at least two reasons for this: first, the discovery in the year 2000 that LAM cells carry TSC2 gene mutations, linking LAM with cellular pathways including the PI3K/Akt/mTOR axis, and allowing the Tuberous Sclerosis Complex (TSC)-regulated pathways that are believed to underlie LAM pathogenesis to be studied in cells, yeast, Drosophila, and mice. A second reason for the rising interest in LAM is the discovery that LAM cells can travel to the lung, including repopulating a donor lung after lung transplantation, despite the fact that LAM cells are histologically benign. This “benign metastasis” underpinning suggests that elucidating LAM pathogenesis will unlock a set of fundamental mechanisms that underlie metastatic potential in the context of a cell that has not yet undergone malignant transformation. Here, we will outline the data supporting the metastatic model of LAM, consider the biochemical and cellular mechanisms that may enable LAM cells to metastasize, including both cell autonomous and non-cell autonomous factors, and highlight a mouse model in which estrogen promotes the metastasis and survival of TSC2-deficient cells in a MEK-dependent manner. We propose a multistep model of LAM cell metastasis that highlights multiple opportunities for therapeutic intervention. Taken together, the metastatic behavior of LAM cells and the involvement of tumor-related signaling pathways lead to optimism that cancer-related paradigms for diagnosis, staging, and therapy will lead to therapeutic breakthroughs for women living with LAM.
Introduction
Genetic Data Support a Metastatic Model of LAM Pathogenesis
LAM can occur in women with tuberous sclerosis complex (TSC), who carry germline TSC1 or TSC2 gene mutations (TSC-LAM), or in women who do not have TSC (sporadic LAM). TSC is an autosomal dominant disorder characterized by seizures, mental retardation, autism, cerebral cortical tubers, subependymal giant cell astrocytomas facial angiofibromas, cardiac rhabdomyomas, renal angiomyolipomas, renal cysts, and LAM. 1 Cystic lung disease consistent with LAM occurs in about 30% of women with TSC, although in most cases it is asymptomatic.2,3 TSC is caused by germline mutations in the TSC1 gene on chromosome 9q34 or the TSC2 gene on chromosome 16p13. 1
Angiomyolipomas are benign tumors found in the kidneys of about 50% of women with the sporadic form of LAM and in the vast majority of patients with TSC. 4 The first evidence of a genetic link between TSC and sporadic LAM was from studies of renal angiomyolipomas from women with the sporadic form of LAM, when it was found that these angiomyolipomas have loss of heterozygosity (LOH) in the TSC2 region of chromosome 16p13. 5 Interestingly, in this first paper, TSC2 LOH was also found in multiple lymph nodes from a woman with extrapulmonary LAM. In retrospect, these genetic findings in extrapulmonary LAM may have been the first clue of LAM cell metastasis (Table 1).
LAM, lymphangioleiomyomatosis.
After identifying TSC2 gene region LOH in sporadic LAM-associated angiomyolipomas, we used single-stand conformation polymorphism analysis (SSCP) to search for mutations in all 41 exons of TSC2. TSC2 mutations (point mutations or small deletions) were identified in five of seven angiomyolipomas from women with sporadic LAM in which TSC2 LOH had been previously found. 6 Some of these were identical to germline mutations that had been previously reported in TSC patients. This work clearly established that inactivation of both alleles was involved in sporadic LAM angiomyolipomas, consistent with the two-hit tumor suppressor gene model, 7 which was originally proposed by Alfred Knudson. Dr. Knudson was an early advocate of TSC and LAM research at Fox Chase Cancer Center in Philadelphia, PA, and working with Raymond Yeung, identified the Tsc2 gene mutation in the Eker rat model, which develops renal carcinomas and uterine leiomyomas. 8
After identifying bi-allelic TSC2 mutations in sporadic LAM-associated angiomyolipomas, laser capture microdissection was used to isolate LAM cells from these patients. TSC2 mutations identical to those in the angiomyolipomas were found in the microdissected LAM cells of four of the patients. 6 The presence of same TSC2 mutation in LAM cells and angiomyolipoma cells, but not in normal kidney, normal lung, or peripheral blood lymphocytes, led to the “benign metastasis” model of LAM pathogenesis. 9 Subsequently, Sato et al. confirmed these findings in Japanese patients with LAM, 10 and Yu et al. demonstrated with the use of multiple microsattelite markers across chromosome 16p13 that the region of LOH appeared to be the same between the angiomyolipomas and the microdissected LAM cells from the same patient. 11
These data were consistent with a metastatic model. However, there were several published case reports of recurrent LAM after lung transplantation in which a woman with LAM had received a male donor lung, and in which Y chromosome fluorescent in situ hybridization (FISH) appeared to indicate that the recurrent LAM cells contained a Y chromosome and therefore arose from the donor lung. To address the genetic origin of recurrent LAM cells more definitively, we studied microdissected recurrent LAM cells from a woman who had received a male-donor lung transplantation, and compared them with her “native” LAM cells prior to the transplantation. The same somatic TSC2 mutation, in exon 18 of TSC2, was present in LAM cells before and after the lung transplantation, 12 thereby establishing that the recurrent LAM cells were genetically related to the original LAM, pretransplantation. In addition, Y-chromosome FISH combined with HMB-45 immunofluorescent staining revealed that her recurrent LAM cells lacked a Y chromosome, consistent with the mutational data and a model in which the recurrent LAM cells arose from her original LAM cells, and not from the male donor lung. In retrospect, in the earlier studies in which it appeared that the recurrent LAM cells contained a Y chromosome, it seems likely that the close interdigitation of LAM cells with reactive stromal cells made the in situ hybridization studies difficult to interpret, a conclusion which Bittman et al. have now validated. 13
LAM Cells Are Present in Blood and Other Body Fluids
The mechanisms through which LAM cells metastasize were mysterious until 2004, when a major finding was made by Crooks et al. (Table 2). 14 Using a density gradient centrifugation separation designed for circulating cancer cell enrichment, they isolated LAM cells from the peripheral blood of women with the sporadic form of LAM. LAM cells, validated by the presence of chromosome 16p13 LOH, were found in blood from 33 of 60 (55%) women with LAM, in urine from 11 of 14 (79%) sporadic LAM patients with angiomyolipomas, and in chylous fluid from one of three (33%) women with LAM. Further work by this group used the expression of surface glycoprotein CD44 and its splice variant CD44V6 to isolate LAM cells from primary cultures derived from explanted lungs of 12 women with the sporadic form of LAM. 15 A great deal remains to be learned about these circulating LAM cells, including their origin, their relationship to pathogenesis, their potential use diagnostically, and their role as a biomarker of disease severity and/or response to therapy.
LAM, lymphangioleiomyomatosis.
Loss of TSC1 and/or TSC2 Enhances Cell Migration and Invasion
The cell autonomous mechanisms underlying the metastasis of LAM cells are likely to include abnormalities of the actin cytoskeleton. Lamb et al. reported that TSC1 expression increases Rho activity, enhances focal adhesion and stress fiber formation via a direct interaction with the ezrin–radixin–moesin family of cytoskeletal proteins. 16 We found that cells lacking Tsc2 have lower levels of Rho activity, enhanced chemotactic cell migration, and decreased cell adhesion. 17 Goncharova et al. 18,19 found that TSC2 regulates cell migration and impacts the activity of both Rac and Rho. Interestingly, they found that the N-terminus of TSC2 was sufficient to inhibit cell migration and activate Rac1, suggesting that this is independent of the GTPase-activiating protein (GAP) activity of tuberin and independent of Rheb.
Expression of Metalloproteinases (MMPs) and Circulation of LAM Cells
Numerous studies have shown that MMPs or regulators of MMPs are expressed in LAM cells. These include expression of MMP-2,20–22 MMP-2 activator, membrane type (MT) MT-1-MMP or MMP-14,22–24 and MMP9. 21 The tissue inhibitors of metalloproteinase TIMP-1, TIMP − 2, and TIMP − 3 were expressed at low level in LAM cells.21,22 Doxycycline treatment to block MMP activity resulted in an improvement in the respiratory function of one patient. 25 A recent study by Lee et al. found that cells derived from a LAM-associated angiomyolipoma exhibited higher of MMP2 activity, and that this was not sensitive to rapamycin, suggesting mTORC1-independence. 26
Cathepsin-K is a cysteine protease that can degrade extracellular matrix (ECM). Chilosi et al. studied 12 cases of pulmonary LAM and found strong immunoreactivity for cathepsin-K in LAM cells in all cases. 27 These studies suggest that expression and/or activation of proteases may contribute to ECM remodeling, facilitating the entry of LAM cells to the circulation. Protease expression and activation may also contribute to cystic degeneration of the lung in LAM patients.
Estrogen Promotes the Survival and Metastasis of Tsc2-null Cells
The role of estrogen in LAM pathogenesis remains a key research question, highlighted by the exceptionally high female predominance of LAM and the possibility of therapeutic benefit of targeting the estrogen axis. Many studies have shown estrogen receptor alpha (ERα) and progesterone receptor (PR) expression by LAM cells,28–32 and our group has found that angiomyolipomas from sporadic LAM patients also express ERα and PR. 31 Howe et al. found that estrogen promotes the proliferation of Tsc2-null ELT3 cells in vitro and the growth of xenograft subcutaneous tumors in vivo. 33 Estrogen treatment of Tsc2-null ELT3 cells or TSC2-null LAM-associated angiomyolipoma cells is associated with strong and rapid (5 min) ERK-1/2 activation, suggesting that estrogen may have cytoplasmic “nongenomic” effects on LAM cells as well as genomic effects.34–36 An interaction between tuberin and the estrogen receptor has been observed,35–37 although its role in LAM pathogenesis remains unknown.
We have studied Tsc2-deficient Eker rat derived ELT3 cells that are estrogen-receptor positive and estrogen responsive, 33 found that estrogen promotes a five-fold increase in the number of pulmonary metastasis in ovariectomized mice carrying subcutaneous tumors. In male mice, estrogen treatment results in a three-fold increase in lung metastasis of ELT3 cells. Estrogen also enhances the number of circulating tumor cells and the survival of intravenously injected ELT3 cells. 38 In vitro, estrogen-treated ELT3 cells exhibit induced resistance to anoikis and decreased levels of caspase-3 cleavage. Using the Xenogen bioluminescent imaging (Caliper Life Sciences, Hopkinton, MA), estrogen treatment causes a two-fold increase in lung colonization of intravenously injected ELT3-luciferase cells at 3 h, and a five-fold increase at 24 h (Fig. 1). Estrogen treatment of the ELT3 cells is associated with activation of MAPK both in vitro and in vivo, and the MEK inhibitor CI-1040 completely blocks estrogen-promoted lung metastasis. 38 These data suggest that a major mechanism underlying the female predominance of LAM may be estrogen-enhanced survival and resistance to anoikis, thereby promoting the survival of circulating LAM cells.
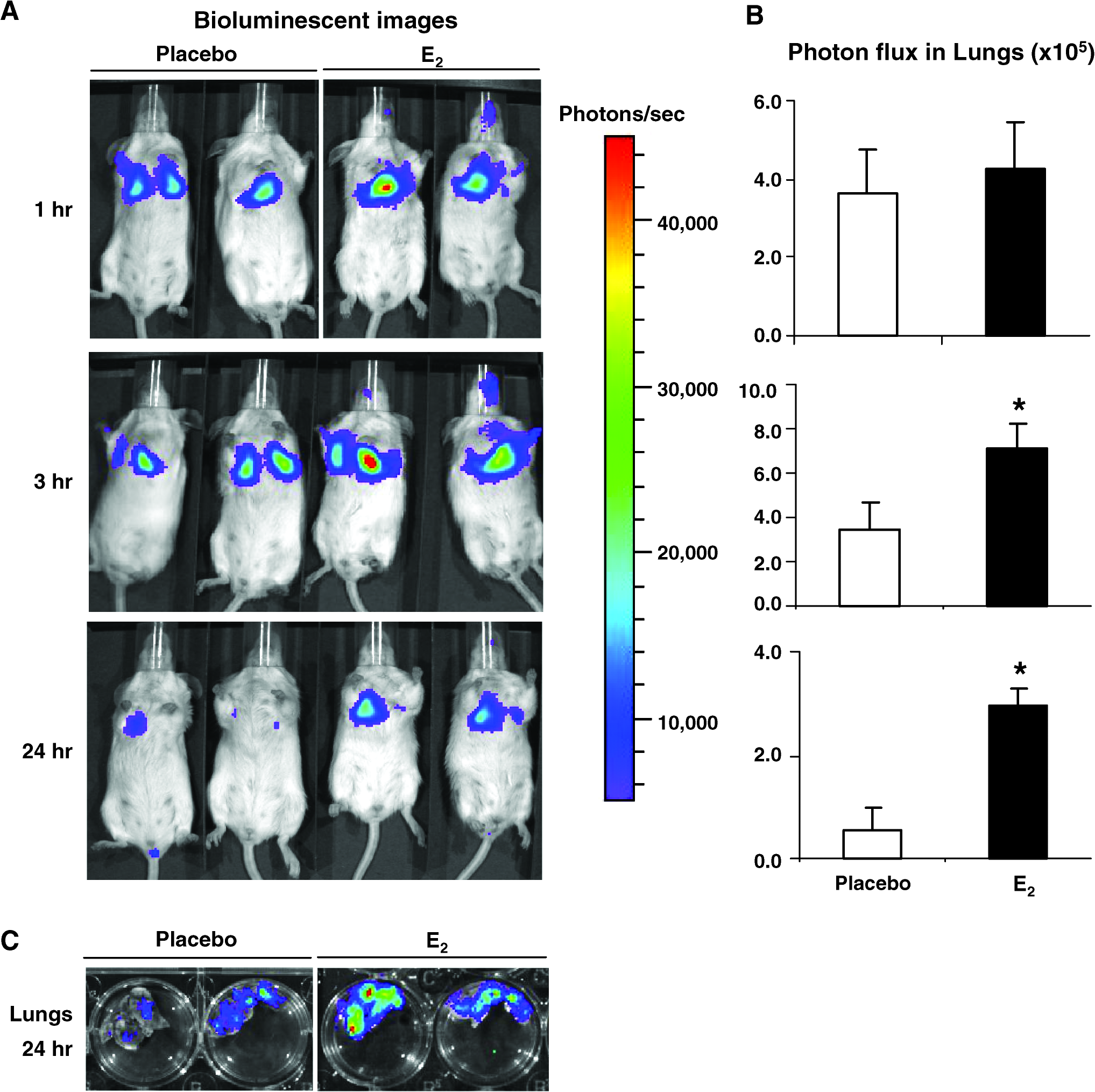
Estrogen promotes the lung colonization of Tsc2-null ELT3 cells. Female ovariectomized CB17-SCID mice were implanted with estradiol (E2) (n = 3) or placebo (n = 3) pellets (2.5 mg, 90-day release) one week prior to cell injection. 2 × 105 ELT3-luciferase cells were injected intravenously. Lung colonization was measured using bioluminescence at 1, 3, and 24 h after injection. (
Role of the Lymphatic System
Kumasaka et al. found that lymphatics were extremely abundant in both pulmonary and extrapulmonary LAM, using a specific marker for lymphatic endothelial cells (LEC) Flt-4 (VEGFR-3). In contrast, only modest evidence of capillary endothelial cells was observed based on CD31 staining. In addition, expression of vascular endothelial growth factor (VEGF)-C was detected in LAM cells, and clusters of LAM cells surrounded by Flt-4 positive cells were observed in the dilated lymphatic vessels, suggesting a specific recruitment of LEC to LAM cells. 39
Subsequently, the same group found LAM cell clusters lined by LEC in chylous fluid, the thoracic duct, and retroperitoneal lymph nodes in five of five LAM patients. 40 These findings suggested that LAM cells are surrounded by and may recruit lymphatic capillaries, providing entry to the lymphatic circulation and promoting metastasis.
Seyama et al. found that the serum level of VEGF-D was significantly higher in 44 LAM patients (average 1069.3 pg/mL) compared to 22 healthy controls (average 295.9 pg/mL). All LAM cells from 44 pulmonary LAM specimens exhibited immunoreactivity for VEGF-D, suggesting that LAM cells are secreting VEGF-D. 41 Glasgow et al. 42 and Young et al 43 also found that VEGF-D levels are higher in LAM patients than healthy volunteers. In addition to its potential role in LAM pathogenesis and as a target for LAM therapy, VEGF-D may be useful diagnostically and/or as a biomarker of disease in LAM patients with lymphatic involvement.
How Can the Metastatic Model Contribute to Early Detection, Biomarker Development, Therapy?
The original “perfect storm” hit the northeast United States on October 31, 1991, driven by three separate weather systems: warm air coming from one direction, cool dry air coming from another, and moisture provided by a hurricane. That same year, a study by Shepherd et al. found that LAM was the fourth most common cause of TSC-associated death. 44 At that point, the TSC genes had not yet been cloned, the genetic links between TSC and sporadic LAM were unknown, and the possibility that LAM cells were metastatic had not been considered. We now believe that LAM is driven by a confluence of prometastic factors that include mTOR activation, lymphatic recruitment, extracellular matrix remodeling, and estrogen promoted cell survival (Fig. 2). Like the perfect storm, even a modest change in any one event contributing to LAM pathogenesis could lessen the overall severity considerably. There are multiple potential sites of therapeutic intervention, including blocking the recruitment of lymphatic vasculature, inhibiting proteases, and inducing apoptosis by inhibiting estrogen or the estrogen receptor.

Multistep model of lymphatic-mediated and estrogen-promoted metastasis of LAM progenitor cells. Genetic studies indicate that LAM progenitor cells carrying TSC1 or TSC2 mutations metastasize to lymph nodes, kidneys, and lungs. Collective studies suggest that LAM cell metastasis is driven by several distinct cellular and molecular mechanisms. We propose a multistep model of lymphatic and estrogen-associated metastais.
Footnotes
Acknowledgments
We thank Peter Mallen (Schepens Eye Research Institute at Harvard Medical School) for the graphic art. This article is dedicated to Alfred Knudson, M.D., Ph.D., Ann Petri, Ph.D., and William A. Petri.
Author Disclosure Statement
No competing financial interests exist.
Research in our laboratory is supported by grants from The Adler Foundation, the LAM Treatment Alliance, the LAM Foundation, and the National Heart, Lung and Blood Institute.