
Abstract
Abstract
The cells comprising pulmonary lymphangioleiomyomatosis (LAM) and renal angiomyolipomas (AMLs) are heterogeneous, with variable mixtures of cells exhibiting differentiation towards smooth muscle, fat, and vessels. Cells grown from LAM and AMLs have likewise tended to be heterogeneous. The discovery that LAM and AMLs contain cells with mutations in the TSC1 or TSC2 genes is allowing investigators to discriminate between “two-hit” cells and neighboring cells, providing insights into disease pathogenesis. In rare cases, it has been possible to derive cells from human tumors, including AMLs and TSC skin tumors that are highly enriched for TSC2-/- cells. Cells derived from an Eker rat uterine leiomyoma (ELT3 cells) are Tsc2-null and these have been used in a rodent cell models for LAM. Further improvements in the ability to reliably grow well-characterized TSC2-/- cells from human tumors are critical to developing in vitro and in vivo model systems for studies of LAM pathogenesis and treatment.
Introduction
Cellular Composition of Lymphangioleiomyomatosis
Lung LAM cells develop within nodular structures as spindle-shaped (proliferative) and epithelioid cells. The spindle-shaped cells express α-smooth muscle actin (SMA), vimentin, desmin, proliferating nuclear antigen (PCNA), and Ki-67. On the other hand, epithelioid LAM cells in addition to expressing smooth muscle antigens, also express melanoma markers such as gp100 (a splice variant of Pmel17), MART-1, CD63, and PNL2. 3 Spindle-shaped and epithelioid cells are immunophenotypically distinct and yet it is possible that they are derived from a common progenitor. A related tumor type, called perivascular epithelioid cell neoplasms (PEComas), is composed of mesenchymal cells, possessing a myomelanocytic phenotype.4,5
LAM nodular structures are coated by hyperplastic type II pneumocytes, which appear proliferative and ultrastructurally have apical microvilli, electron-dense lamellar bodies, and cytoplasmic projections. 6 The hyperplastic nature of the type II pneumocytes may be related to the expression of thyroid transcription factor-1 which regulates, in part, synthesis of surfactant proteins.7,8 The role of these cells in the pathogenesis of LAM is poorly understood.
Lymphatic endothelial cells and mast cells are also major cellular constituents of LAM lung nodules. Lymphatic endothelial cells (LEC) were identified by their reactivity with anti-VEGFR-3 and anti-podoplanin antibodies. 9 Numerous lymphatic channels lined by lymphatic endothelial cells are found within LAM lung nodules. LEC may surround LAM cells in LAM cell clusters, which are seen in LAM chylous effusions. 9 Mast cells within the LAM nodule were identified by reactivity with anti-CD117 antibodies, which recognizes c-Kit, a 145-kD transmembrane protein that is highly expressed by mast cells. 10
LAM Cells
With multiple cell types comprising LAM nodules, it appears that the interplay of these cells, as well as cells in neighboring tissues, is involved in the pathogenesis of LAM. However, only LAM cells appear to have TSC gene mutations. The mutations found most frequently in sporadic LAM inactivate the TSC2 tumor suppressor gene.11–13
Lung LAM cells have been grown from explants of lung tissue obtained at the time of transplant and from diagnostic biopsies. LAM cells have been grown in different culture systems directly from explants or following enzymatic digestion. A major challenge is the finding that all approaches yield a heterogeneous population of cells. All cells grown from explants of LAM lung do not show loss of heterozygosity (LOH) at the TSC2 locus, suggesting that these cultures contain LAM cells mixed with other cells lacking the genetic features of LAM cells. Consistent with this interpretation, subpopulations of cells are immunoreactive with antibodies to SMA and gp100, and some nuclei show allelic deletion of the TSC2 gene (Fig. 1). Methods are being developed to isolate and propagate pure populations of LAM cells. When populations are isolated from heterogeneous cultures using fluorescence-activated cell sorting, LOH or allelic imbalance for TSC2 is observed mostly in cells positive for the cell surface marker CD44v6 (Fig. 2). Unfortunately, the same antibody used to isolate these cells also induces cell death, blocking efforts to grow a pure population of cells. 14

Phenotypic and genotypic heterogeneity in LAM cell cultures. Reaction of cells cultured from LAM lung (
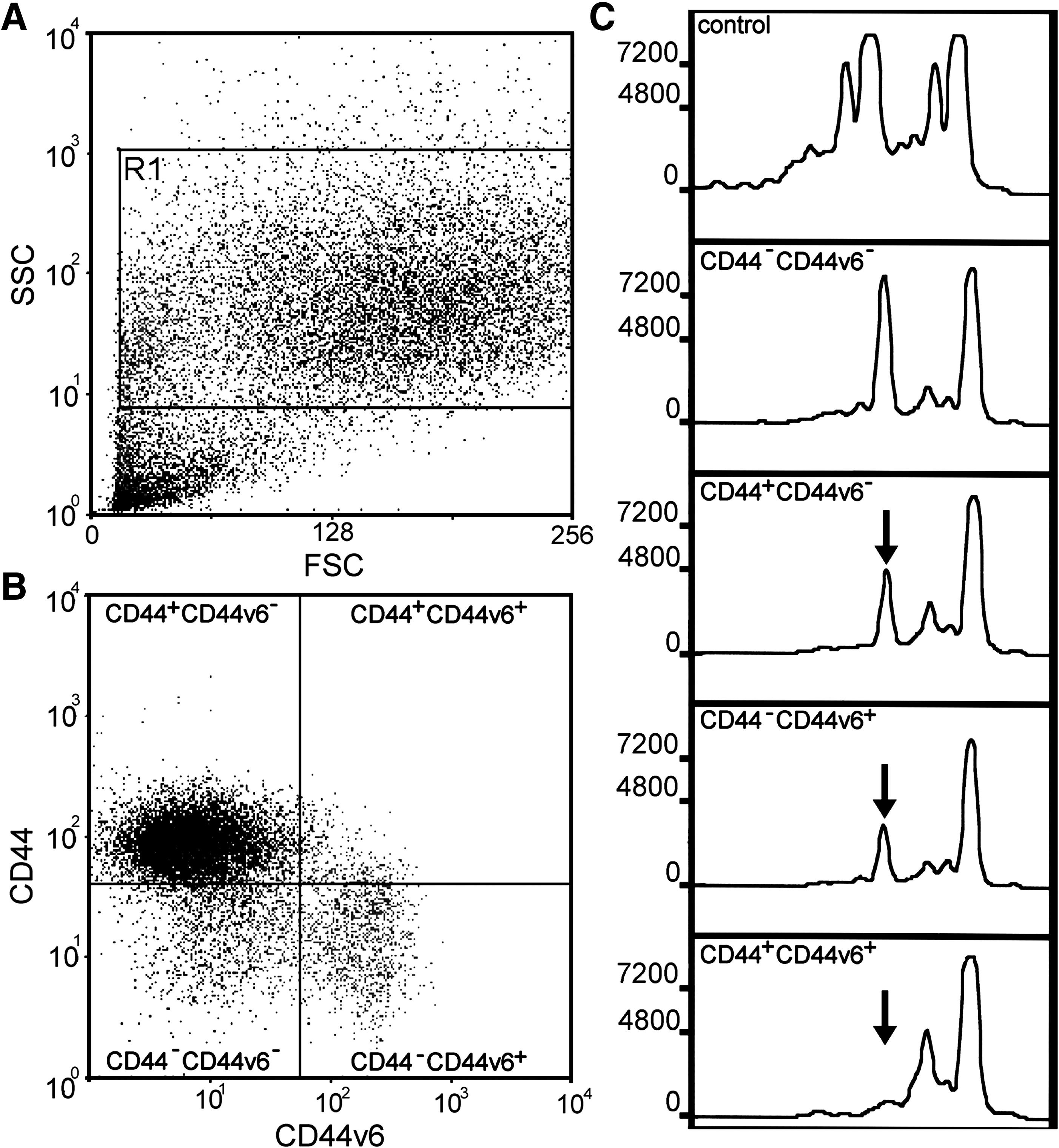
Enrichment of LAM cells showing loss of heterozygosity (LOH) at the TSC2 locus. Cells were incubated with CD44-R-phycoerythin and CD44v6-fluorescein antibodies for fluorescence-activated cell sorting. (
Loss of TSC2 protein function results in hyperactivation of the mTORC1 signaling pathway leading to dysregulated cell growth, and biochemical events related to mTORC1 activation have been used to characterize cells derived from LAM nodules. Cells derived from LAM nodules exhibit hyperphosphorylation of p70S6K and ribosomal protein S6, 15 markers commonly used to assess mTORC1 activation. Hyperphosphorylation of S6 cannot be relied upon exclusively as a marker for LAM cells, however, because its presence does not always equate with loss of TSC2 function. For example, malignant cells from melanoma often posses hyperactivated S6 but without TSC2 mutations. 16 Furthermore, S6 phosphorylation may result from activation of phospholipase D and protein kinase C, 17 and low expression of DEPTOR could result in activation of mTOR. 18 The potential for hyperphosphorylation of S6, independent of mutations inTSC2, supports the scientific concern that LAM cells should be genetically characterized. In addition, it may be necessary to characterize the cells epigenetically, since methylation of the TSC2 promoter has been shown to play a role in AMLs. 19
Multiple mitogens and growth signaling pathways may drive LAM cell proliferation. Among these mitogens are angiotensinogen II, 10 insulin-like growth factor, 20 INF-γ and INF-β,21,22 plasminogen, 23 prostaglandins, 24 and PDGF. 25 Interestingly, angiotensin II could also upregulate VEGF production. Altogether, these data suggest that the microenvironment of the LAM cell may generate factors responsible for their proliferation.
Sporadic LAM affects adult women almost exclusively, suggesting that tumor growth is hormonally regulated. Lung LAM cells express estrogen and progesterone receptors26–28 and patients treated with tamoxifen and progesterone showed downregulation of these respective receptors. 29 In mice xenografted with Tsc2-null cells derived from Eker rats, 17-β-estradiol increased metastasis in both male and female mice. 30 Estrogen also enhanced the growth of liver hemangiomas in Tsc mice. 31 Consistent with hormonal effects on LAM cells, it has been suggested that estrogens induce metalloprotease production by on LAM cells, which affects their invasive properties. 32 Although all of these findings appear congruent with the pathogenesis of lung LAM, a clear effect of estrogens on lung LAM cells cannot be concluded because of the lack of genetic information on cells that have been studied and the use of non-human cell lines.
LAM cells are histologically benign, so it was surprising when evidence began to accumulate that LAM cells were able to metastasize. Although the initial site of LAM and the cell of origin have not been identified, transplanted lungs showed recurrence of LAM due to invasion by LAM cells derived from the recipient.33–37 This behavior is in agreement with the finding of disseminated LAM cells in the blood and lymphatic circulation. 38 Thus, an important challenge is to define molecular aspects of LAM cell metastasis.
CD44v6 is expressed by many cancer cells and appears to be important for metastasis. It is expressed by LAM lung lesions, as well as in cultured cells grown from LAM lungs (Fig. 2). 14 CD44v6 splice variant interacts with integrins and multiple receptors (e.g., hepatocyte growth factor receptor). The multiple interactions of CD44 and its splice variants suggest that it may participate in cell-to-cell interactions within the LAM nodule microenvironment and with the extracellular matrix.
It has been suggested that LAM cell invasiveness and migration are due to the activation of the GTPases Rac1 and Rho, which are, in part, regulated by TSC225,39 and that estrogens drive the synthesis of metalloproteases by LAM cells. 32 These studies were carried out with cells obtained from LAM lungs treated with collagenase D and elastase. 15 The cells obtained express TSC2 and contain high levels of phosphorylated S6. As suggested by these authors, the use of these type of cell cultures present limitations due to the heterogeneity. 25 Therefore, it is difficult to conclude that the LAM cells lacking TSC2 function are responsible for invasion and migration and that they respond to specific stimuli. To study cell signaling pathways in LAM cells, it would be advantageous to use a homogeneous population of human LAM cells with mutations in TSC2.
Global gene expression analysis of microdissected LAM lung cells of patients with sporadic LAM identified a set of transcripts that distinguished lung LAM cells from smooth muscle and melanoma cells (GEO database GSE12027). Some chemokines were differentially expressed and may participate in attraction and anchoring of cells to sites of metastasis. In agreement, CCL2/MCP-1 was increased in bronchoalveolar lavage fluid of LAM patients compared to healthy volunteers. Furthermore, LAM cells within the lung lesions expressed a distinct group of chemokine receptors, including CCR2, CXCR4, CCR7, and CCR10. It was also observed that CCL2/MCP-1 selectively mobilized cells from explants of lungs of LAM patients with TSC2 LOH and that CCL2 could participate in a positive feedback loop of metastasis. 40
In summary, obtaining LAM cells that are clonal or highly enriched has been challenging due to the multiple cell types derived from lungs and the heterogeneity of LAM nodules. To understand the role of signaling pathways in LAM pathogenesis and response to treatment, LAM cells that are well characterized genetically, phenotypically, and functionally would be of use to the LAM scientific community.
Cellular Composition of Angiomyolipomas (AMLs)
AMLs were first described as large renal tumors comprised of fat, muscle, and blood vessels. 41 Renal AMLs may occur in otherwise healthy individuals or in association with TSC or sporadic LAM. Up to 80% of TSC patients eventually develop AMLs, and women develop them more frequently than men. As in LAM, this may suggest hormonal regulation of growth, and AMLs often express receptors for both estrogen and progesterone.42,43 Smooth muscle cells of pulmonary LAM and AMLs appear to be closely related histologically, immunohistochemically, and in some cases genetically, suggesting a model in which renal AML cells migrate or metastasize to the lung, resulting in pulmonary LAM. 38 Metastases of angiomyolipoma cells to the lung may cause development of LAM.44,45 AMLs sustain second-hit mutations in either TSC1 or TSC2, as indicated by LOH analysis.46,47
The histologically distinct cell types comprising AMLs appear to be derived from perivascular epithelioid cells capable of differentiating into cells with features of melanocytes, smooth muscle, and fat.48,49 In addition, AMLs contain multiple vessels types, four of which (cellular, hemangiopericytic, glomeruloid, and aneurysmatic) have loss of heterozygosity (LOH), indicating that they are neoplastic while one (collagenous) does not have LOH. 50 LOH of vessels, smooth muscle, and fat cells is consistent with the hypothesis that AMLs are derived from a mesenchymal precursor that retains the ability to differentiate into multiple different lineages.
Cells from AMLs
In 2001, Arbiser et al. described the generation of cultures of human AML cells, carrying phenotypic characteristics of cells derived from AMLs. 51 The stable cell line was obtained by sequentially introducing SV40 large T antigen and human telomerase into tumor cells from a sporadic human AML of a patient not affected by TSC. In another study, the cells of a primary culture were isolated from a LAM-associated renal angiomyolipoma. These cells carried inactivating mutations in both alleles of the TSC2 gene (TSC2 exon 16 missense mutation G1832A) and expressed estrogen receptor α, estrogen receptor β, and androgen receptor. 52 Exposure of these cells to estradiol and tamoxifen citrate promoted their growth. 52 Tamoxifen acts as an estrogen agonist in these cells, in contrast to tamoxifen's estrogen antagonist action in Eker rat-derived ELT-3 cells. 53 The stimulated growth was associated with phosphorylation of p42/44 mitogen-activated protein kinase (MAPK) and increased expression of c-myc, indicating an activation of nongenomic and genomic signaling pathways, respectively, in cultured angiomyolipoma cells. 52 Consistent with the observation that cells lacking tuberin have constitutive hyperphosphorylation of p70S6K and its ribosomal substrate S6, these cells showed increased S6 phosphorylation.15,17,54 In LAM-associated and sporadic AMLs, the mTOR p70S6K signaling pathway is highly activated in relation to TSC1/TSC2 mutation. 55 The limitation of this cell model was the number of studies since the entire primary culture was lost after a few passages.
Two cell populations were described in 2005 that were derived from an AML of a TSC2 female patient. 56 These cell populations were isolated, characterized, and separated by sequential subcloning in order to obtain homogeneous cultures. One cell population had a flat and elongated appearance and these cells were alpha actin-positive smooth muscle-like cells (ASM), while the other had an epithelial-like morphology and were keratin 8/18-positive. Both populations were reactive to HMB-45, a monoclonal antibody that recognizes LAM lung cells.14,31 The epithelial-like cells were markedly reactive with anti-RhoA antibodies. Both cell populations contained a germline TSC2 exon 18 mutation, consisting of a base-pair change in the codon for amino acid 698, leading to replacement of lysine with a stop codon (K698X). Only smooth muscle-like cells exhibited LOH and did not express tuberin (TSC2-/- ASM cells), whereas in the epithelial-like cells the mutation was heterozygous and the cells expressed tuberin. LOH in TSC1 or TSC2 has been documented in AMLs, cardiac rabdomyomas, and LAM cells, and only rarely in cerebral cortical tubers and skin lesions.11,47,57,58
More recently, another alpha actin-positive cell population was isolated and characterized, this time from an AML of a male patient with a germline mutation in TSC2. 19 These cells were reactive with HMB45 and anti-CD44v6 antibodies, while tuberin was undetectable. These cells bear a germline mutation in TSC2 at the junction between intron 8 and exon 9, but they lack LOH. The loss of tuberin expression is the consequence of methylation of the TSC2 promoter (TSC2-/meth ASM cells), suggesting that angiomyolipoma pathogenesis might also originate from epigenetic defects. The involvement of methylation in the inhibition of the TSC2 gene has been confirmed by expression of tuberin following treatment with chromatin-remodeling agents such as trichostatin A and 5-azacytidine. 19 Moreover, these agents downregulate the constitutive S6 phosphorylation. Methylation of promoter regions is a common feature in human cancer.
In spite of the different genetic mutation, TSC2-/- and TSC2-/meth ASM cells share proliferative and biochemical characteristics. Their growth and proliferation required epidermal growth factor (EGF) that cannot be replaced by insulin like-growth factor 1 (IGF-1) (Fig. 3A).19,56 TSC2-deficient cells secrete IGF-1, which may act as a survival factor by promoting the expression of survivin. 59 The EGF-dependent growth is related to the lack of tuberin since transfection of the TSC2 gene in TSC2-/- ASM cells and the treatment of TSC2-/meth ASM cells with 5-azacytidine reduced their proliferation to a normal rate, decreased S6 phosphorylation and eliminated the requirement of EGF for growth (Figs. 3B, 3C, and 3D).19,60 Blockade of either EGF or IGF-1 receptors with specific antibodies resulted in cell death (Fig. 3A).19,56 The EGF receptor signaling pathway regulates cell differentiation, proliferation, migration, angiogenesis, and apoptosis, all of which become deregulated in cancer cells. Anti-EGF receptor antibody has been shown to be efficacious in several types of cancer, such as colorectal and head and neck cancers 61 and it may also be of therapeutic value in TSC.

(
Rapamycin has been recently reported to be effective in promoting the regression of TSC AMLs. 62 The exposure of TSC2-/- ASM cells to rapamycin resulted in a cytostatic action only when the drug was added at the time of plating the cells, otherwise the drug was rather ineffective. TSC2-/meth ASM cells are more responsive to rapamycin and the cytostatic effect is observed at any time. These data are consistent with the effect of rapamycin in a large trial that reported the tendency of AMLs to increase in volume after therapy was stopped. 62 In accordance with these results, rapamycin was highly effective in suppressing the growth of Tsc mouse kidney cystadenomas but a marked tumor regrowth occurred after stopping the drug. 63 The exposure to rapamycin and anti-EGF receptor antibody downregulated the constitutive S6 phosphorylation observed in TSC2-/- and TSC2-/meth ASM cells.15,54 Growth factors, such as EGF, lead to Erk-1 and Erk-2 phosphorylation through the activation of MAPK that regulates tuberin. Both rapamycin and anti-EGF receptor antibody inhibited Erk phosphorylation with anti-EGFR receptor antibody being more effective. All together these data support the regulatory role of MAPK pathway in TSC2 ASM cell proliferation.
Cellular Composition of TSC skin tumors
Patients with TSC develop multiple benign skin tumors that are located on the face (angiofibromas and forehead plaques), back (shagreen patch), and nails (ungual fibromas). TSC skin tumors may be present at birth, but they typically develop throughout childhood, appearing as small red papules or larger irregular plaques. 64 They are composed mostly of fibrous tissue, and fibroblast-like cells are a major cell population. These fibroblast-like cells (variously called fibroblasts, fibroblastic cells, interstitial fibroblasts, and stroma cells) have been the focus of most studies of TSC skin tumors.
In early histological studies of angiofibromas and ungual fibromas, investigators observed that fibroblast-like cells are larger than normal fibroblasts, stellate or polygonal in shape, and sometimes multinuclear.65–67 Fibroblast-like cells are interspersed among increased amounts of collagen and dilated vessels, including blood and lymphatic endothelial cells.68,69 The epithelium overlying the tumors is often thickened due to a proliferation of keratinocytes. 70 Once the genetic basis for TSC was uncovered and antibodies were generated against human TSC1 and TSC2 proteins, it was possible to test for aberrant expression of these proteins in TSC tumors. Immunohistochemistry revealed that a significant proportion of TSC skin tumors showed loss of TSC1 and/or TSC2 expression in the interstitial fibroblast-like cells.71,72 Other cellular constituents, such as vessels and keratinocytes, did not show loss of TSC1 or TSC2 expression.
Loss of TSC1 or TSC2 expression in the fibroblast-like cells suggested that these cells had sustained second-hit mutations in either TSC1 or TSC2. Attempts to demonstrate a second-hit mutation in TSC skin tumors were mostly negative, and loss of heterozygosity (LOH) at the TSC1 or TSC2 locus was observed in only a few TSC skin tumors.47,58,73–75 Inability to detect LOH in most samples was likely due to the cellular heterogeneity of these tumors. This problem was surmounted by using fluorescence in situ hybridization to analyze individual nuclei, which documented allelic deletion for TSC2 in dermal touch preparations of 8/8 angiofibromas and 6/7 periungual fibromas. 70 These results indicate that fibroblast-like cells are the two-hit cells in TSC skin tumors.
Loss of TSC1–TSC2 function in fibroblast-like cells is expected to lead to activation of mTORC1 and hyperphosphorylation of ribosomal protein S6. When sections of TSC skin tumors were stained for phospho-S6, immunoreactivity was observed in the fibroblast-like cells in the dermis as expected (Fig. 4A), whereas fibroblasts in normal-appearing skin were negative (Fig. 4B). 70 Surprisingly, the epidermis overlying tumors also stained positive for phospho-S6. Keratinocytes did not show genetic alterations at the TSC2 locus, and positive staining in the epidermis may be caused by the release of paracrine factors from fibroblast-like tumor cells. 70 The potential for paracrine activation of phospho-S6 represents a potential pitfall in using phospho-S6 as the sole marker for tumor cells.
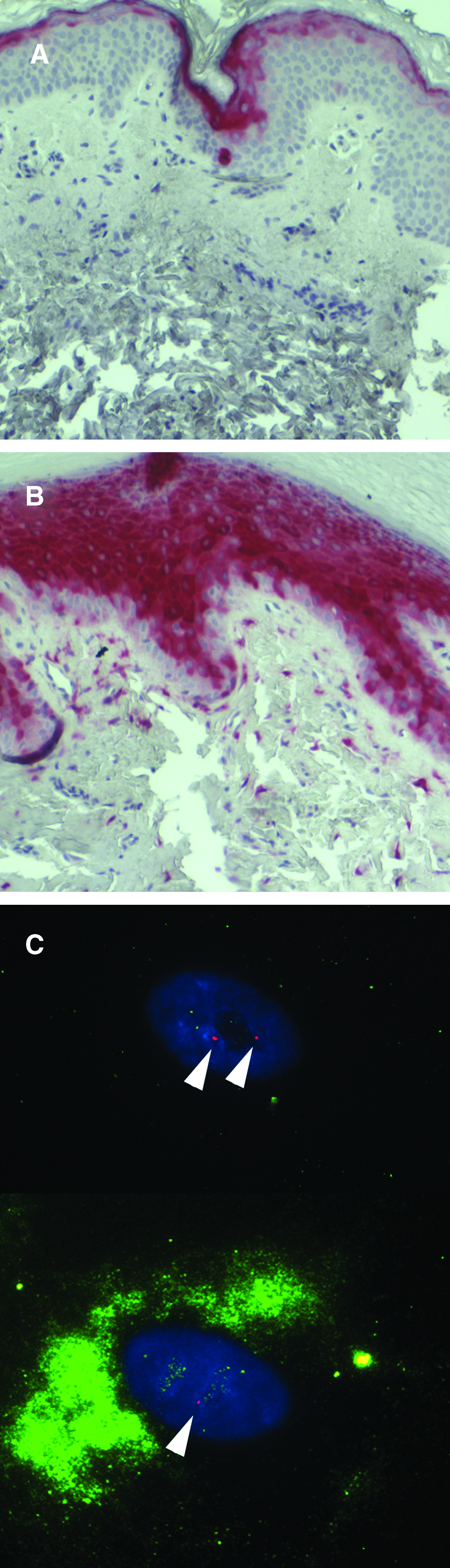
Heterogeneity of mTORC1 activation in TSC skin tumors and cells grown from TSC skin tumors. Sections of normal-appearing skin (
Cells from TSC Skin Tumors
Fibroblast-like cells can be grown from tissue explants of TSC skin tumors. Nearly all cultures of angiofibromas and ungual fibromas contain two-hit cells, but they generally comprise a minority of the cells 70 and cultures do not show LOH. 76 This heterogeneity is similar to that typically observed when cells are grown from explanted LAM lung nodules. 14 Heterogeneity in skin tumor cultures was further dissected by combining immunocytochemistry for phospho-S6 with fluorescence in situ hybridization for deletion of TSC2. Individual cells were observed that were phospho-S6 negative with 2 signals for TSC2, and phospho-S6 positive with 1 signal for TSC2 (Fig. 4C). These results are consistent with mixture of one-hit with two-hit cells, with mTORC1 activation in the two-hit cells. In rare instances, cells were grown that exhibited biallelic mutations in TSC2, undetectable TSC2 protein, and constitutive activation of mTORC1. 77 These TSC2-/- cells will be useful for future studies on the effects of loss of TSC2 in TSC skin tumors, but most studies to date have used tumor-derived cells that are either genetically uncharacterized or heterogeneous with one-hit and two-hit cells.
Cells grown from angiofibromas and ungual fibromas express fibroblast markers such as HSP47 and vimentin, but are immunonegative for SMA, S-100, CD14, and CD68.78,79 Whereas their immunophenotype is similar to fibroblasts, they exhibit several differences with normal fibroblasts. In one study, angiofibroma cells displayed reduced proliferation and increased senescence compared to normal fibroblasts, 80 while another study found comparable levels of BrdU incorporation, a prolonged S phase, and increased apoptosis. 76 These results are difficult to reconcile with studies showing that loss of function of the TSC1–TSC2 complex results in increased proliferation, 81 but it may be related to a phenomenon observed in hematopoietic stem cells whereby deletion of TSC1 produces transient expansion of stem cells, followed by loss of long-term repopulating potential. 82
Fibroblast-like cells grown from TSC skin tumors have been used to investigate potential mechanisms of tumorigenesis. TSC skin tumors are highly angiogenic, 68 and loss of TSC2 function results in increased expression of the angiogenic cytokine VEGF.83,84 Gene expression arrays were used to identify additional growth factors or chemokines that might play roles in TSC skin tumorigenesis. Two genes that are highly overexpressed by fibroblast-like cells are monocyte chemoattractant protein-1 (MCP-1, or CCL2) 79 and epiregulin. 70 MCP-1 is a chemokine that attracts peripheral blood monocytes. It is also highly expressed in skin of transgenic mice, having a dominant negative allele of Tsc2. 85 Overexpression of MCP-1 in TSC skin tumors is associated with recruitment of mononuclear phagocytes into the tumors, where they may further stimulate angiogenesis. 79 Epiregulin is an epidermal growth factor (EGF)-family member that may induce changes in the epithelium overlying TSC skin tumors. 70
Tsc2-/- Cells from Animal Tumors
Several rodent models of Tsc deficiency have been developed, and murine cell lines established from these animals are widely available. Like bone fide LAM cells, for which these lines are often used as a surrogate, these cells lack either Tsc1 or Tsc2. As a result, they recapitulate the signaling defect caused by loss of Tsc in LAM cells. Specifically, these lines exhibit upregulation of mTORC1 signaling and downregulation of AKT activity. This shared signaling defect has made these cells useful in preclinical studies, where they have proven their utility for evaluating efficacy of targeted therapies for LAM, such as mTORC1 inhibition using rapamycin and its analogs.
Cells Derived from Knockout Mice
Germline inactivation of both alleles of the Tsc1 or Tsc2 tumor suppressor gene in mice is embryonic lethal.86–89 However, mouse embryo fibroblasts (MEFs) have been isolated from both Tsc1 and Tsc2 knockout mice. Interestingly, establishment of Tsc-null MEF lines in culture requires inactivation of p53, which was achieved by crossing Tsc heterozygous mice with p53 heterozygotes.87,90 The requirement that p53 be lost to establish Tsc-deficient MEFs in culture suggests that loss of Tsc function results, directly or indirectly, in dysregulation of this tumor suppressor, and that abrogation of p53 function(s) is required to overcome senescence and/or induce immortalization in this setting of Tsc deficiency. Like tumors that arise in Tsc-deficient animals, and LAM lesions, Tsc-null MEFs exhibit upregulation of mTORC1 signaling. Recently, MEF cell lines have also been isolated from mice expressing a hypomorphic Tsc2 allele (Tsc2del3/del3), and these also display activated mTORC1 signaling. 91
In addition to MEF cell lines, a line of neuroepithelial progenitor (NEP) has also been established from Tsc2-null mice. 92 These cells exhibit an aberrant differentiation pattern resembling giant cells of human tumors and express β-III tubulin and GFAP. Like Tsc2-null MEFs, Tsc2-null NEP also exhibit upregulation of mTORC1 signaling.
Interestingly, in addition to downregulation of AKT, cell lines lacking Tsc also exhibit decreased IRS1 and PDGFR activity, thought to be due to feedback loops associated with constitutive upregulation of mTORC190,93,94 and/or the requirement for Tsc2 as a co-factor for the kinase activity of mTORC2, which is responsible for AKT phosphorylation at S473. 95
Cells Derived from Tsc-Null Tumors
The most widely used cell lines are derived from tumors that arose in the Eker rat model for tuberous sclerosis. Eker rats are heterozygous for Tsc2,96,97 and are susceptible to spontaneous and carcinogen-induced tumors in the kidney (renal cell carcinoma:RCC),98,99 uterus (uterine leiomyoma), 100 as well as spleen and highly vascular areas of the skin (hemangiosarcoma).101,102 These rats also develop pituitary and brain lesions.103,104 Several cell lines have been isolated from spontaneous tumors of Eker rats. Lines have been derived from RCCs from these animals, termed ERC lines, 99 and from neoplastic/polycystic kidneys that develop in these animals when loss of the wild-type Tsc2 allele occurs during development, the EKT lines. 105 In addition to these tumor-derived cell lines, lines have also been isolated from Eker rat embryos that are Tsc2-null (EEF-8) or Tsc2-proficient (EEF-4). 106
However, the cell lines most often utilized as surrogates for LAM are the proliferative smooth muscle cell lines isolated from Eker rat uterine leiomyomas, the ELT cell lines. 107 Like the proliferative smooth muscle cells of LAM lesions, these lines are Tsc2-null and have constitutively active mTORC1 signaling. 108 More importantly, these lines express estrogen and progesterone receptors, and retain their hormone responsiveness.109,110 Isogenic ELT3 cell lines, T3 and V3, re-expressing Tsc2 or vector control, respectively, have also been generated. 111 Recently, it has been demonstrated that in response to estrogen, ELT cells grown as subcutaneous tumors in SCID mice will migrate to the lung. 30 This important finding could potentially lead to the development of an exciting new in vitro/in vivo animal model for LAM.
Preclinical Studies Using Tsc-Deficient Cell Lines
As mentioned above, both Tsc1- and Tsc2-deficient cell lines exhibit upregulation of mTORC1 signaling. Not surprisingly, growth of these cell lines in vitro and as tumors in nude mice is inhibited by rapamycin and its analogs. The elevated S6K and S6 phosphorylation characteristic of dysregulated mTORC1 signaling in Tsc/p53-null MEFs is inhibited by rapamycin, 87 as is their growth in culture. 90 ELT smooth muscle cells are also inhibited by rapalogs in vitro. 108 Growth inhibition by rapamycin reflects what is seen in vivo, where growth of Tsc-deficient tumor cells is inhibited by rapamycin and its analogs in several mouse models, including nude mice bearing subcutaneous tumors derived from injection of Tsc/p53-null MEFs. 112 Similarly, tumor development in Eker rats is inhibited by rapamycin, as is the growth of ELT3 cells in SCID mice.30,54,108
Conclusions
The cellular microenvironments of LAM, AMLs, and TSC skin tumors are complex, with multiple cell–cell and cell–matrix interactions. It comes as no surprise that cells grown from these tumors also tend to be heterogeneous. The recent availability of methods to distinguish different cell populations based on genetic or molecular markers is providing new insights into disease pathogenesis and response to treatment. Further improvements in methodologies are needed to be able to reliably isolate and grow pure populations of LAM cells, as well as other cell populations comprising these tumors, in order to more accurately assess their relative contributions to tumor formation. This will allow the development and testing of new therapies that specifically target LAM cells or the interactions of LAM cells with neighboring cells or matrix.
Footnotes
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, National Heart, Lung, and Blood Institute, and TS080064 from the Congressionally Directed Medical Research Program (TND). The work of A. Gorio and E. Lesma was supported by the Ministero Superiore della Sanita (IRCCS Grant 2007–09 on rare diseases program), the Italian TSC Association, and the Italian Lymphangioleiomyomatosis Association. We thank the Tuberous Sclerosis Alliance and The LAM Foundation for patient referral. We thank Dr. Martha Vaughan for useful discussions and critical review of the manuscript.
Author Disclosure Statement
Drs. Darling, Pacheco–Rodriguez, Gorio, Lesma, Walker, and Moss have no conflicts of interest or financial ties to disclose.