
Abstract
Abstract
Pediatric hepatic angiosarcoma is a very rare malignant vascular tumor. A few cases have shown pediatric hepatic angiosarcoma occurring on a background of preexisting vascular lesions. We report the case of a newborn girl who presented extensive limbs and upper trunk cutaneous mixed vascular malformations at birth. These malformations were associated with thrombocytopenia. Cutaneous biopsies revealed complex vascular malformations with a significant lymphatic component. Compressive body suit therapy led to regression of the limbs' cutaneous vascular malformations. At the age of 9 months, the patient presented multiple heterogeneous hepatosplenic nodules. Aggressive treatment with prednisone, vincristine, and hepatosplenic embolizations resulted in initial improvement of the hepatosplenic lesions for few months, followed by an increase of the lesions with failure of response to treatment despite adding alpha-interferon-2b to treatment. The patient died at the age of 19 months. The autopsy's pathological examination revealed a hepatic-based angiosarcoma with plurimetastatic dissemination to the spleen, lungs, peritoneum, pleura, mesenteric linings as well as the serosa of the stomach and small intestine. Multiple cutaneous and visceral complex capillaro-lymphatico-venous malformations were also identified. We hypothesize that these multiple extensive mixed vascular malformations were associated with chronic lymphedema which probably predisposed to the development of the angiosarcoma in our patient.
Introduction
Case report
A newborn girl presented at birth with extensive cutaneous vascular lesions located on the upper extremities and upper trunk associated to thrombocytopenia (platelets at 27.109/L) (Fig. 1). The initial diagnosis was kaposiform hemangioendothelioma without full-blown Kasabach–Meritt syndrome. Cutaneous biopsies revealed mixed vascular malformations demonstrating a significant lymphatic component (Fig. 2). There was no histological evidence of kaposiform/epithelioid hemangioendothelioma or infantile hemangioma. Abdominal ultrasound was normal, there was no gastrointestinal bleeding and no additional evidence of vascular disease in the rest of the body. Treatment with a short course of oral prednisone and compressive bodysuit therapy led to resolution of the thrombocytopenia as well as remarkable regression of the limbs' vascular malformations (Fig. 3).
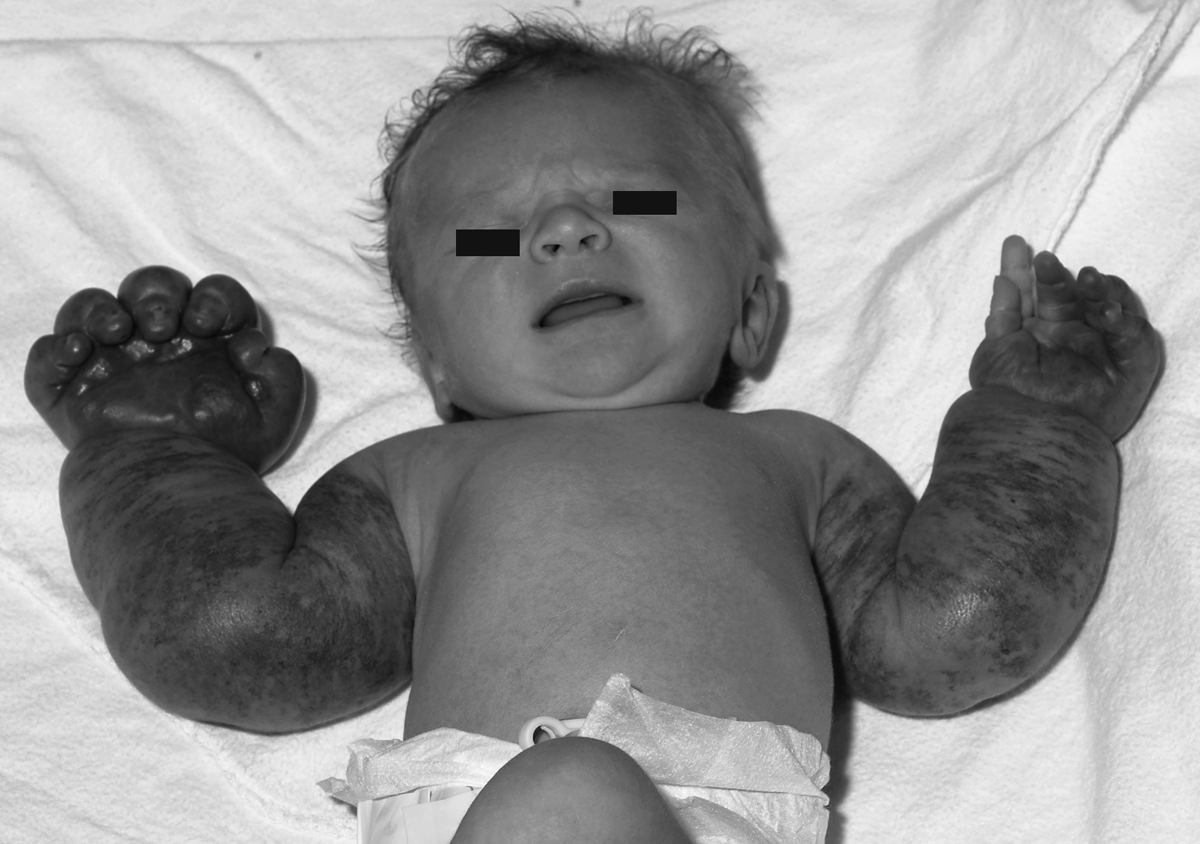
Bilateral extensive complex vascular malformations involving upper extremities at birth.
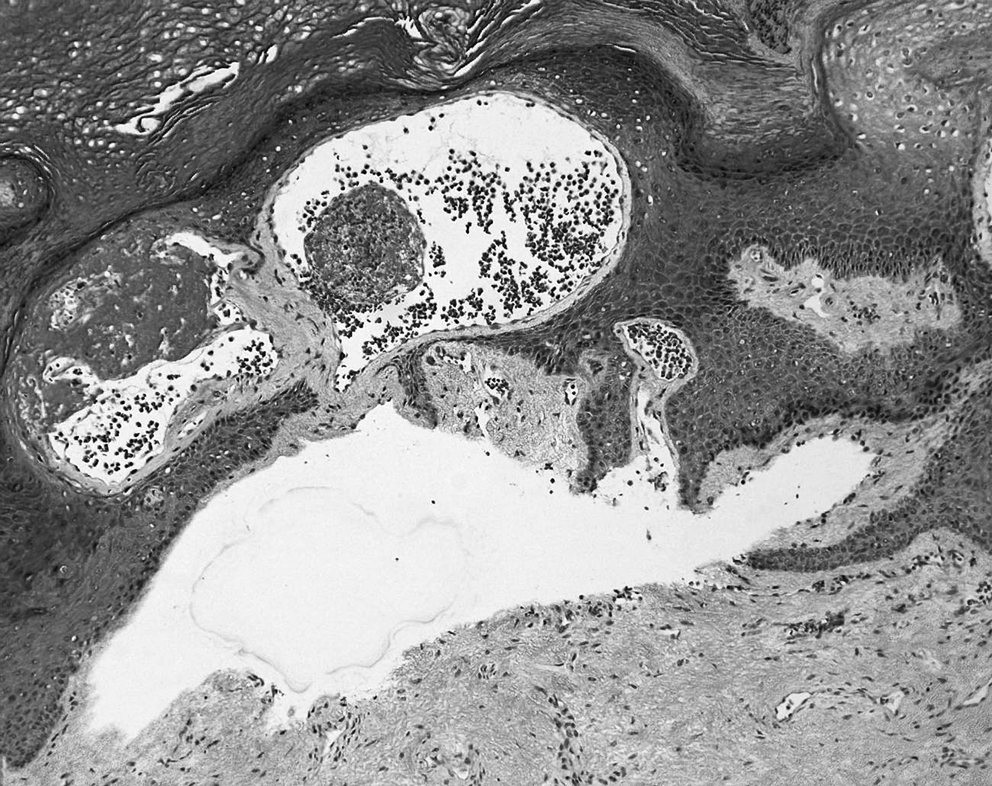
Complex vascular malformations presenting a significant lymphatic component, D2-40 ×200.

The patient at 8 months of age with remarkable regression of the cutaneous vascular malformations following compressive bodysuit therapy.
At 9 months of age, the patient presented with diarrhea, irritability, loss of appetite, and hepatosplenomegaly. Clinically, the limbs and trunk's vascular malformations were unchanged. There was a recurrence of the thrombocytopenia (platelets at 25.109/L). Moreover she had a normocytic anemia (Hb = 105 g/L), increased fibrinogen (4.98 g/L, Normal values: 2.00–4.00 g/L) and positive D-dimers. Liver and renal function tests as well as α-fetoprotein were normal. Abdominal ultrasound, CT scan, and MRI revealed enlarged liver and spleen with multiple heterogeneous hepatosplenic nodules. The gallium scan was normal. Both radiologically-guided liver biopsy and a repeated cutaneous biopsy revealed complex vascular malformations with significant lymphatic component. There were no histological or immunohistochemical findings suggestive of infantile hemangioma or hemangioendothelioma. Bone marrow biopsy was normal. Treatment with prednisone, vincristine, and hepatosplenic embolizations resulted in remarkable improvement of the hepatosplenic lesions at 14 months of age.
However, at 16 months of age, the patient presented ascites with radiologically documented increase of the size of the multifocal hepatosplenic nodules. Moreover, multiple bilateral lung nodules were found on pulmonary CT scan. Treatment with prednisone, vincristine, and alpha-interferon-2b failed. The child died at 19 months of age due to respiratory failure.
At autopsy, the pathological examination revealed multiple cutaneous and visceral capillaro-lymphatico-venous malformations associated with a hepatic-based lymphangiosarcoma presenting plurimetastatic dissemination to the spleen, lungs, peritoneum, pleura, mesenteric linings, as well as the serosa of the stomach and small intestine. In the liver, the histological picture consisted of irregular anastomosing vascular channels varying greatly in size and shape and lined with atypical endothelial cells. The latter presented polymorphic, hyperchromatic nuclei with prominent nucleoli and a high mitotic index (more than 5 mitoses per mm2) (Fig. 4). Foci of more discrete angulated blood vessels resembling patch stage Kaposi's sarcoma (Fig. 5) as well as more solid zones of atypical spindle cells resembling plaque stage Kaposi's sarcoma (Fig. 6) were visualized. Numerous foci of vascular invasion and emboli were identified, especially within the liver parenchyma. Multiple zones of necrosis were seen. Intertwined and adjacent to the hepatic angiosarcoma, foci of malformative capillary, venous and lymphatic blood vessels were readily identified (Fig. 7). Immunohistochemistry revealed that malignant vascular endothelial cells were positive for endothelial markers CD31 and CD34 (focally), lymphatic marker D2-40, WT-1, and focally for Glut-1. The proliferation index was very high with positive Ki-67 in 85% of endothelial nuclei.
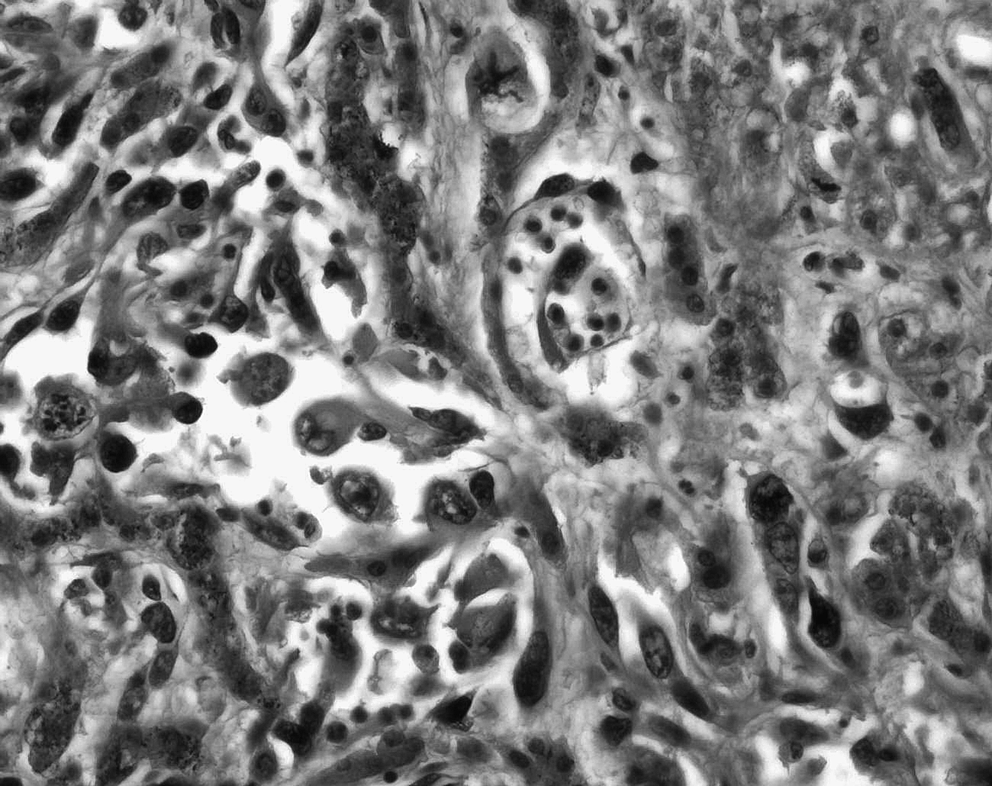
Irregular anastomosing vascular channels varying greatly in size and shape and lined with polymorphous atypical endothelial cells, HPS ×400.
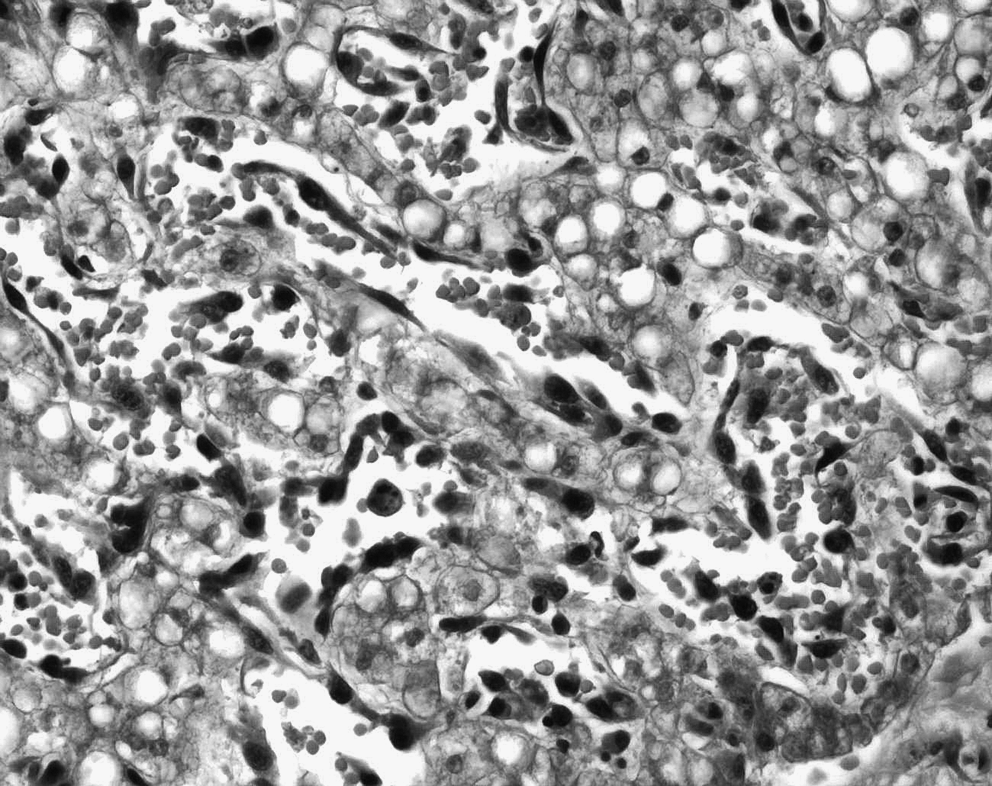
Angiosarcoma with foci of more discrete angulated atypical blood vessels resembling patch stage Kaposi's sarcoma, HPS ×400.
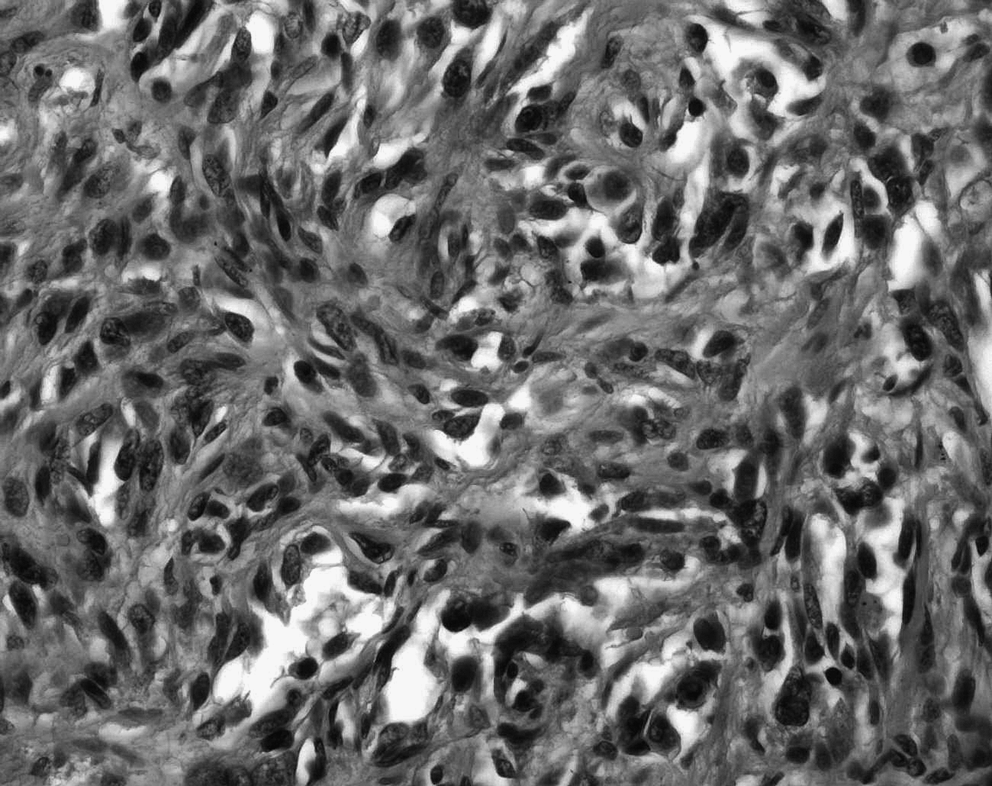
Angiosarcoma with cellular zones of atypical spindle cells resembling plaque stage Kaposi's sarcoma, HPS ×400.
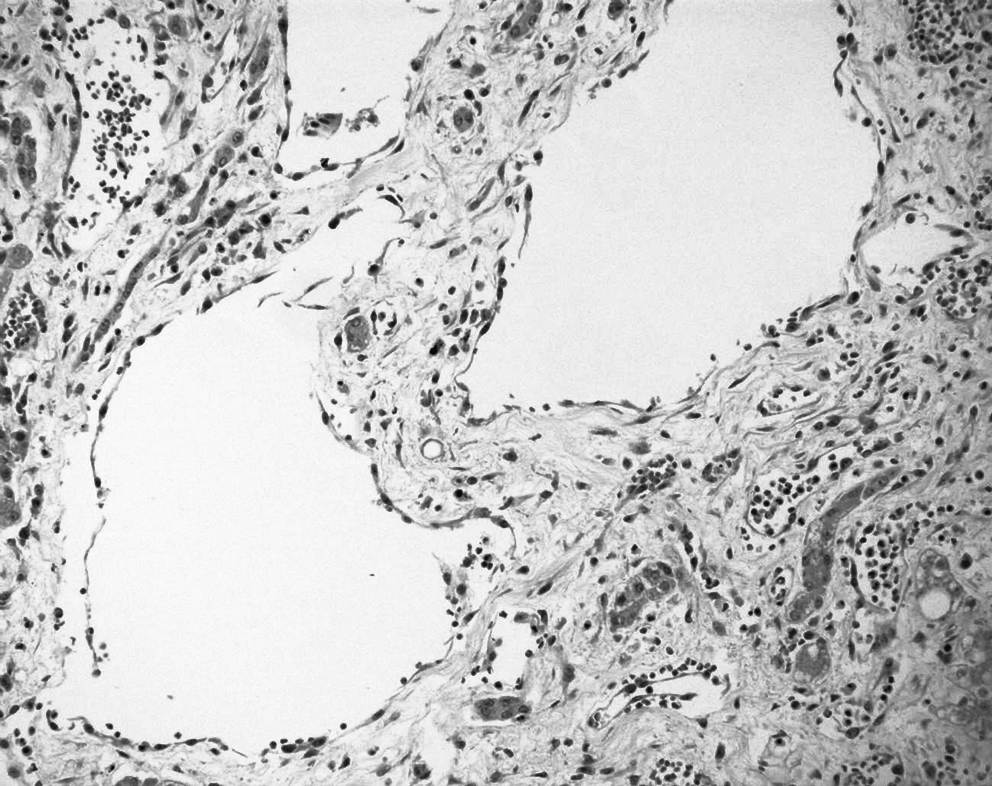
Malformative capillary, venous, and lymphatic blood vessels intertwined and adjacent to the hepatic angiosarcoma, HPS ×200.
Discussion
Angiosarcomas can occur in any region of the body, but most of them arise in the skin and superficial soft tissue, the head-neck region being the most common site of origin. Angiosarcomas of deep soft tissue or with a visceral location are very uncommon. 1 The mean age at presentation in childhood is 40 months. In contrast to hepatic angiosarcoma of the adult, pediatric hepatic angiosarcoma affects females more than males, with a female to male ratio of approximately 2:1 and the age at presentation ranges from 2 months to 15 years (mean age of 3.7 years).4–6
Angiosarcomas have been associated with several preexisting vascular lesions. Girard et al. described three cases of angiosarcoma occurring in port-wine stains. 7 Cutaneous angiosarcoma and pediatric hepatic angiosarcoma have been previously described in the background of infantile hemangiomas.8,9 Pediatric hepatic angiosarcoma has also been reported to develop 4 to 5 years after a primary diagnosis of type 2 infantile hemangioendothelioma.4,10 In these cases, the angiosarcoma may have developed as a result of a transformation or a new lesion. 4 In our case, one could theorize that the hepatic-based angiosarcoma could have resulted from the transformation of a hepatic hemangioendothelioma. However, liver biopsy as well as autopsy findings did not reveal any pathological evidence of hepatic hemangioendothelioma. Moreover, the terminology of infantile hepatic hemangioendothelioma, traditionally used to describe tumors with both benign- and malignant-appearing histological features, has recently fallen into disfavor. 9 In 1971, Denher and Ishak 11 proposed the subclassification of infantile hepatic hemangioendotheliomas into two histologic groups. 9 Type 1 infantile hemangioendothelioma was used to describe the more common subtype that involutes and regresses.5,9 Type 2 infantile hemangioendothelioma is more aggressive, often multicentric, and can be metastatic. 5 However, this subclassification was abandoned by several multidisciplinary centers specialized in vascular anomalies, which consider that type 1 hemangioendotheliomas are rather benign infantile hemangiomas and type 2 hemangioendotheliomas are rather pediatric hepatic angiosarcoma.4,9,12
The association between angiosarcoma and previous radiotherapy, chronic lymphedema, or environmental toxins are well known. 1 Pediatric hepatic angiosarcoma does not have a clear association with environmental carcinogens such as Thorotrast, arsenic, and vinyl chloride. 4 In 1948, Stewart and Treves reported six cases of angiosarcoma in adult patients treated for breast cancer who had postmastectomy lymphedema. They found that angiosarcomas appeared at an average of 12.5 years after radical mastectomy. 10 Stewart–Treves syndrome is now applied more broadly to angiosarcoma resulting from chronic lymphedema secondary to trauma, infection, radiotherapy, or idiopathic causes. Cutaneous angiosarcoma has also been described in the background of radiation and other vascular lesions. 2 To our knowledge, there has been no previous report in the medical literature of an association between a pediatric lymphangiosarcoma and extensive vascular malformations, such as in our case. Although the pathogenesis of angiosarcoma is not known, various mechanisms have been proposed in relation to lymphedema. 13 Stewart and Treves suggested that a systemic carcinogenic factor possibly contributed to both the initial breast carcinoma and the subsequent angiosarcoma. 10 More recently, others have postulated that malignant transformation may occur during the formation of collateral vessels in a lymphedematous area. A third theory proposes that malignancy within lymphedematous sites may be more capable of escaping immune system surveillance. 13 The cutaneovisceral vascular malformations, presenting a significant lymphatic component, could have led to chronic lymphedema in our patient, which probably predisposed to the occurrence of angiosarcoma.
Hepatic angiosarcoma usually presents with rapid abdominal enlargement and jaundice. Other signs and symptoms include abdominal pain, vomiting, fever, tachypnea, dyspnea, periumbilical tenderness, and anemia.4,5 By the time pediatric hepatic angiosarcoma is diagnosed, the lesion is often unresectable and usually involves both hepatic lobes. Metastases are common, frequently to the lungs, such as in our patient, but can also occur in the lymph nodes, pleura, bone, and adrenal gland.4,5,9 The mortality rate for pediatric hepatic angiosarcoma is high. Pediatric dermatologists are often involved in the care of patients with hepatic vascular lesions due to the frequent coexistence of hepatic infantile hemangiomas with cutaneous infantile hemangiomas. Proliferation of hepatic masses after the first year is less characteristic of benign infantile hemangiomas and should raise suspicion of a malignant tumor. 9 Occasionally, in such cases, multiple liver biopsies may be required for definitive diagnosis due to the multifocal distribution of angiosarcoma in association with infantile hemangiomas. However, liver biopsies of vascular lesions are sometimes avoided because of risks related to bleeding. 9 Radiologic studies may play a role in the evaluation of these patients, but frequently there are no distinct radiological features to differentiate malignant from benign vascular tumors.4,13 In addition to the histological features of adult angiosarcoma, pediatric hepatic angiosarcoma may have hypercellular whorls of sarcomatous cells, or “kaposiform” spindle cells similar to kaposiform hemangioendothelioma of soft tissue, such as in our case. A whorl-like arrangement of cells, or glomeruloid foci, can also be seen. Eosinophilic globules are occasionally present in the cytoplasm of the spindle cells in these kaposiform areas. These globules are periodic acid-Schiff (PAS) positive, resistant to diastase digestion, and do not express alpha-1-antitrypsin or alpha-1-antichymotrypsin. Similar globules have not been seen in epithelioid hemangioendothelioma, cavernous hemangioma, or adult hepatic angiosarcoma. Vascular invasion can occur in pediatric hepatic angiosarcoma and mitosis can vary from few to many. 4 Immunohistochemically, these kaposiform areas stain with CD31 and CD34 antibodies, but not with von Willebrand factor.4,13 Moreover, other immunohistochemical stains such as Factor VIII-related antigen, and Ulex europaeus lectin 1 may be useful endothelial markers to help identify angiosarcomas.2,13
Because of their rarity, the optimal management for malignant vascular tumors, especially in childhood, has not been defined yet. Angiosarcomas are confirmed as being aggressive neoplasms with a poor prognosis: their behavior in childhood is not different from their adult counterpart. 1 Average survival of pediatric hepatic angiosarcoma appears to be 16 months. To date, surgery, chemotherapy, radiotherapy, and transplantation have not improved the poor prognosis with only few reported survivors. 4 Complete surgical resection remains the mainstay of treatment, but is probably not sufficient even in cases of small lesions. Radiotherapy may have a significant role in local control and could be combined with surgery, though the tumor site and the age of the child must be considered when radiotherapy is proposed in the case of completely resected tumors. The role of chemotherapy, especially in an adjuvant setting, is uncertain, but the poor outcome in these patients might support the use of chemotherapy and the evaluation of new intensive regimens or new drugs, such as angiostatin receptor annexin II.1,14
Footnotes
Author Disclosure Statement
The authors have no conflicts of interest or financial ties to disclose.