
Abstract
Abstract
Lymphangiomatosis is a rare proliferative disorder of the lymphatic system. The etiology is unknown, rendering it difficult to manage. This case report of lymphangiomatosis with features of Gorham's disease reveals the progressive and unexpected nature of the condition. It highlights the need for further research into the pathophysiology and management of lymphangiomatosis as current treatment options are limited.
Introduction
Clinical Presentation
A Caucasian female child presented to Dermatology at the age of 18 months with a congenital vascular malformation of the anterior left thigh and a recent history of left thigh and buttock swelling. Two months after the onset of swelling she developed intermittent chylous discharge from the cutaneous malformation of her left thigh. The swelling and birthmark continued to extend over the following 12 months, eventually involving the entire left buttock, left thigh, and labia majorum. She had no past medical history of note apart from one urinary tract infection at the age of 2 years.
The swelling and chylous discharge progressed and she was referred to the Lymphovascular Malformation Clinic in 2001, at the age of 3 years. On examination, she had a predominately subcutaneous diffuse cavernous lymphangioma involving the left labium majora and extending over the left inguinal region and hip, spreading down to the middle of the left thigh, and laterally into the left buttock (Fig. 1). The lower limbs were of equal length and there were no clinical signs of venous hypertension.
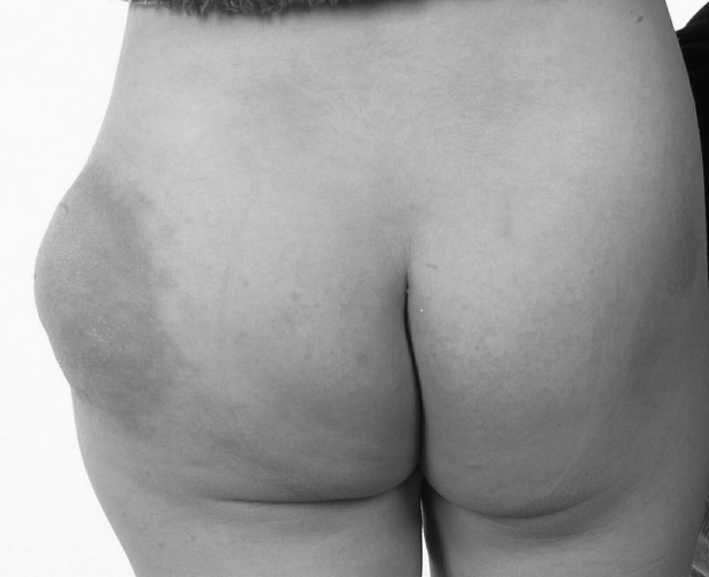
Posterior view of the left hip/buttock demonstrating a lymphatic malformation and discoloration of the overlying skin, aged 3 years.
Investigations
An MRI scan of the pelvis confirmed the presence of an extensive lymphatic abnormality within the subcutaneous tissues lateral and anterior to the left thigh. A network of tiny subcutaneous collections of lymph was seen within the lateral left thigh extending anteriorly through the left inguinal canal along the line of the iliac vessels and the lower IVC to the level of the L3 vertebra. The lymphatic abnormality crossed the midline and extended to the right common iliac vessels and up to the level of L5. The underlying muscles appeared intact. The bladder was deviated to the right and its left wall was indented by the malformation, but the wall itself remained intact. There was no evidence of ureteric dilatation. High signal changes were seen within the L4 vertebral body, and the lymphatic malformation also surrounded the body of L5. The MRI findings supported the clinical suspicion of an extensive lymphatic abnormality with diffuse lymphangiomatosis considered the most likely diagnosis at this stage.
Disease Progression
The lymphatic malformation continued to progress slowly in terms of cutaneous involvement and left thigh/buttock swelling. The patient suffered repeated episodes of cellulitis associated with intermittent chylous discharge from the cutaneous component acting as the portal of entry of infection. She also developed several urinary tract infections as a result of the malformation interfering with the bladder and preventing complete emptying. Low dose prophylactic antibiotics provided intermittent reprieve from both forms of infection. However, the antibiotic of choice would vary every 6–12 months because of a reduction inefficacy. A medium chain triglyceride (MCT) diet was implemented in a bid to reduce the chylous discharge from the lower limb. However, the diet was poorly tolerated at this stage.
Further imaging revealed a lymphocutaneous fistula within the left thigh region, in association with obstructed iliac lymphatic routes. In 2004 and 2005, she underwent lymphatic bypass surgery and achieved a good result with a reduction in size of the malformation over left buttock, and reduction in leakage of chyle. However, the swelling continued to extend across the mid-line to the right groin and down the left leg, presumably as a result of opening up of other smaller lymphatic channels/“cul-de-sacs” after the major ones had been removed surgically.
Complications of Disease Progression
In 2006, aged 8, the patient was referred to the pediatric rheumatologists as she had developed intermittent pains within her lower back. Her symptoms improved with nonsteroidal anti-inflammatory drugs (NSAIDs), but she developed asthma as an adverse event and had to limit the quantity of analgesics that she could consume. An urgent MRI scan confirmed widespread abnormal marrow signal within the vertebral bodies of T7, T8, T10, T12, L1–L5, and the entire sacrum. Multiple pathological compression fractures of all the lumbar vertebrae were present, accounting for her pain. There was no evidence of compromise of the spinal canal. Extensive cystic material was seen in the retroperitoneum encasing the great vessels and extending up into chest through the diaphragmatic hiatus. In the pelvis, the abnormal tissue encased the urinary bladder and rectum. There was extensive involvement of the left gluteal region and groin. Extensive involvement of the bones of the pelvis was noted. There was also involvement of the left femoral neck and proximal shaft. Comparisons were made with previous films, and the overall conclusion was of an extensive lymphangiomatosis with features of Gorham's disease.
At this stage she was commenced on bisphosphonates and the MCT diet was reinstated. The diet appeared to have a positive impact on the frequency and volume of chylous discharge from the left lower limb. However, it was more difficult to assess the efficacy of the bisphosphonates. Nonetheless, she continues to receive three-monthly infusions to this date in case they are of benefit.
In 2007, at the age of 9, the patient suffered pelvic collapse due to bony infiltration by the lymphatic abnormality. She became wheelchair-dependent as she was unable to weight-bear on the left lower limb. In April 2008 she underwent cementation of the pelvis by the orthopedic surgeons. Although she suffered a partial femoral nerve injury during the procedure, she had a good surgical result. The bone cementing procedure provided her with pelvic stability and enabled her to weight-bear after several months of rehabilitation. No further orthopedic surgery was offered as it was deemed too complicated, extensive, and “unpredictable.” Similarly, adjuvant radiotherapy was considered too dangerous in view of the extent of the disease, the associated effects on her growth, and the risk of producing a secondary cancer.
Current Disease Status and Management
In July 2010, the patient began to experience neck and back pain. Her mobility began to decline from being able to walk with the aid of crutches to being wheelchair-bound once more. She complained of constant pain in the left hip and was aware of recurrent blood-stained chylous discharge from the left thigh area (Figs. 2 and 3), which was unresponsive to the MCT diet. She had also developed vaginal chylous discharge within recent months. Urgent MRI scans revealed extension of the lymphatic abnormality into the neck, over the top of the lungs, and following the neurovascular bundles of the brachial plexus into the arm. She suffered with pain in her right shoulder and scapula that has affected her ability to write and play wheelchair basketball, the latter skill being highly important to her.
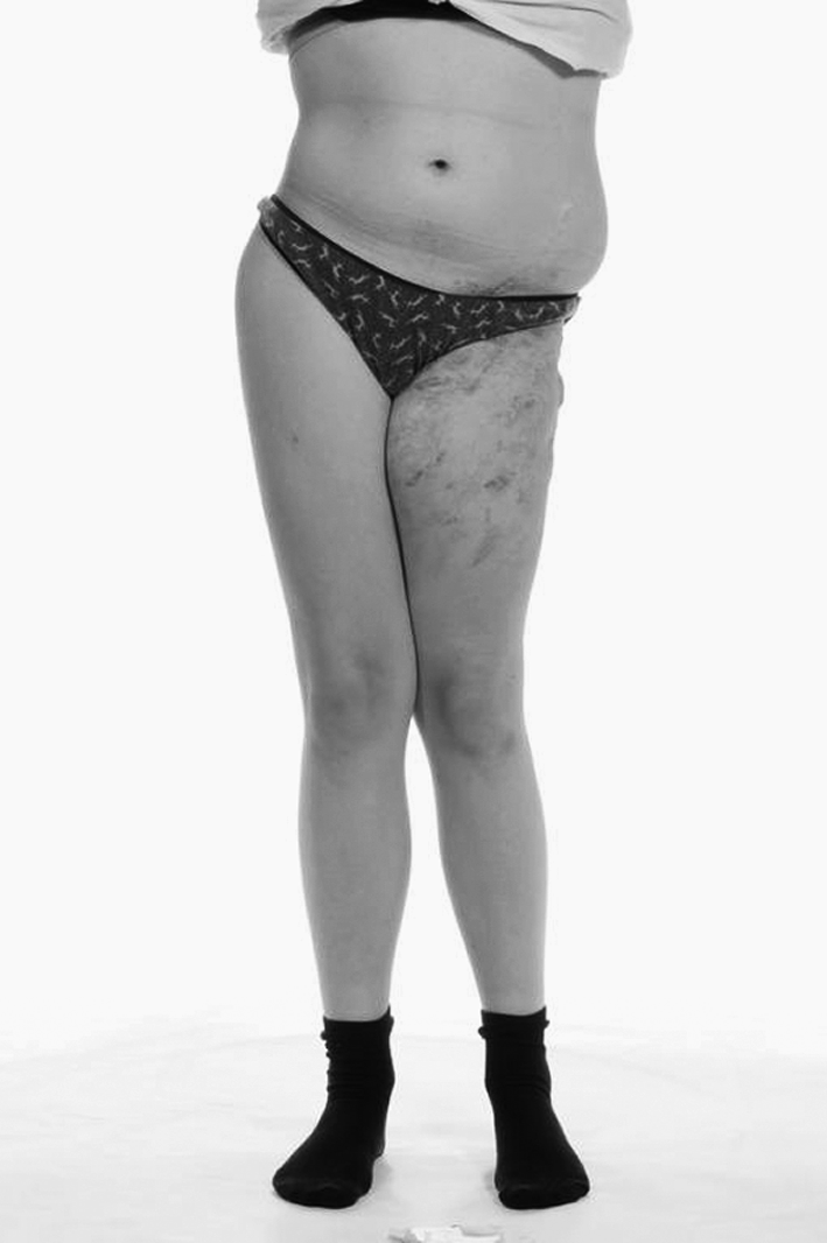
Anterior view of the extensive lymphatic malformation affecting the left flank, pelvis, and lower limb. The bony destruction from Gorham's disease is evident by the abnormal stance of the patient, aged 13 years.
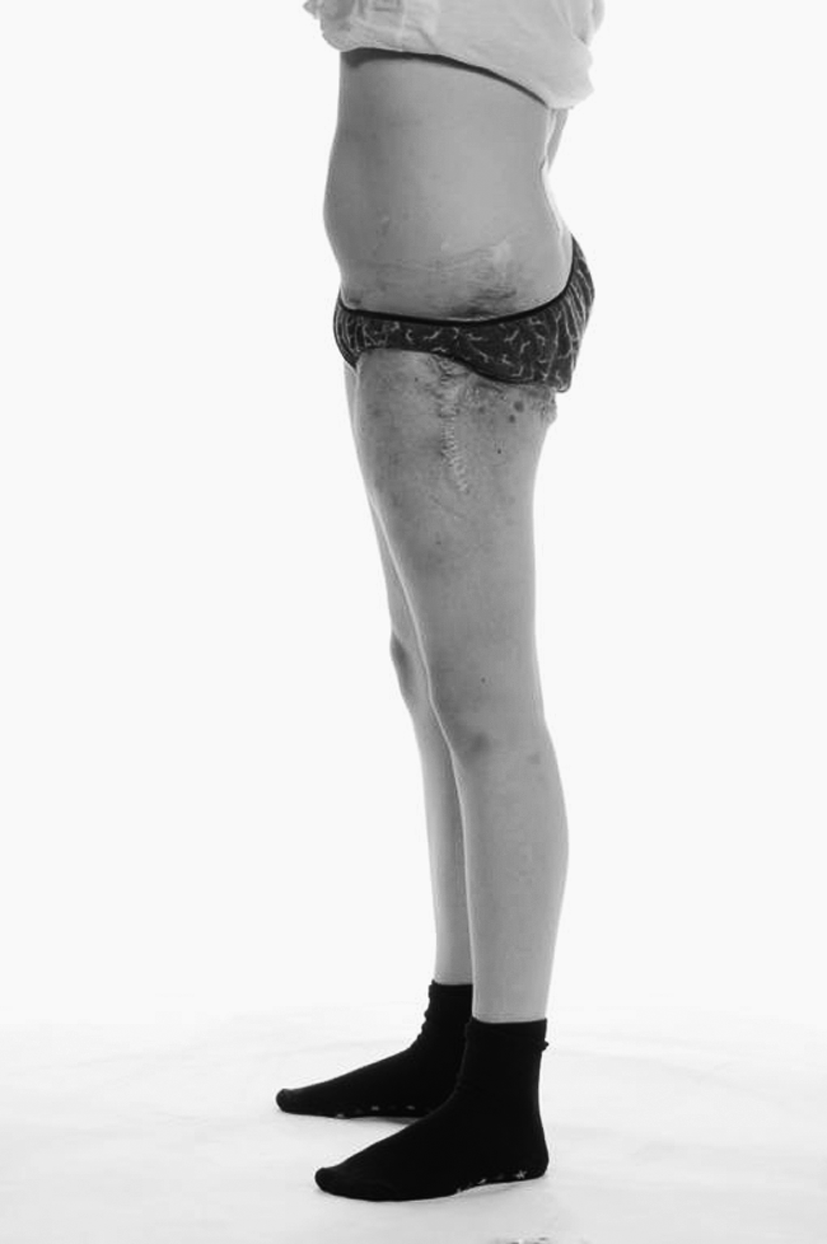
Lateral view of the lymphatic malformation of the left hip. Multiple surgical scars are present, along with numerous lymphangiectasia that frequently leak chyle.
Given the progressive nature of her condition, it was necessary to consider all therapeutic options. A multi-disciplinary team discussion settled on a trial of sirolimus. In 2011, at 13 years of age, the patient was commenced on oral sirolimus titrated to a goal level of 10–15 ng/ml. The intention is that it will halt the progression of her proliferative lymphatic disorder which is slowly infiltrating soft tissues and destroying bone. Hopefully it may stop the chylous leakage, reduce pain, and prevent further bone destruction. We continue to monitor the patient closely to assess her response to treatment.
Discussion
There exists some confusion within the literature regarding the terms “lymphangioma,” “lymphatic malformation,” and “lymphangiomatosis.” Lymphatic malformations represent a failure of lymphatic development and frequently exist in isolation with no communication with otherwise normal lymph drainage pathways. 2 It is best to consider lymphatic malformations as genetically determined structural anomalies with normal endothelial cell turnover, rather than as tumors that are proliferative with endothelial hyperplasia. Malformations increase in size by distension or hypertrophy, not through increased cell division, unlike lymphangiomas. 2
Lymphangiomatosis is a rare disorder of unknown etiology characterized by the proliferation of lymphatic vessels. It has a variable presentation, as a number of different sites may be affected, either in combination or in isolation. These include the dermis, soft tissues, bone and viscera. 1 Lymphangiomatosis of the soft tissues typically presents with a poorly-defined soft, spongy brown-to-purple lesion. 3 Lymphangiomatosis typically presents at birth but may occur in children and young adults.2,4 The clinical outcome of patients with a diffuse lymphangiomatosis is related to the extent of the disease. Visceral involvement (in particular the pleura and lungs) may be associated with a poor prognosis, compared to cases where disease is confined to soft tissue and/or bony involvement. 5
Patients with bony involvement of lymphangiomatosis may be given an additional diagnosis of Gorham's disease (synonyms include Gorham-Stout syndrome, vanishing bone disease, massive osteolysis) 6 or the bony involvement may just be considered part of the lymphangiomatosis process. There appears to be considerable overlap between the two conditions; it is more likely they represent the clinical spectrum of the pathological process of lymphatic proliferation. Patients with Gorham's disease typically present with pain of the affected site, but swelling and deformity may also occur. Gorham's disease occurs when massive osteolysis occurs as a result of infiltration by proliferating vessels. Bruch-Gerharz et al. described capillary, venous, and lymphatic vascular malformative processes occurring within the bony tissues. 7 However, recent studies suggest that the major proliferative component of Gorham's disease is lymphatic in origin. 8 This contrasts Gorham and Stout's original hypothesis of the disease occurring as a result of abnormal proliferating blood vessels. 9
The etiology of lymphangiomatosis and Gorham's disease is poorly understood. It has been proposed that trauma-induced proliferation of vascular granulation tissue may underlie some cases of Gorham's disease. 9 Interleukin-6 (IL-6)-mediated increased osteoclast activity has been suggested as another mechanism for Gorham's but fails to explain the more diffuse forms of lymphangiomatosis. 10 Lastly, vascular endothelial growth factors have been implicated in lymphangiogenesis.11,12 Recent advances made in understanding the pathways of lymphangiogenesis are likely to be relevant to the aforementioned diseases. Platelet-derived growth factor (PDGF) signaling pathways are involved in angiogenesis, 13 and recently PDGF-BB has been implicated as a lymphangiogenic growth factor. 14 VEGFR-3 and LYVE-1 have been shown to be involved in lymphatic development. 15 It has been proposed that the disordered lymphangiogenesis seen in Gorham's disease may be the result of disruptions in these pathways. 16 Further understanding of these pathways may ultimately lead to the development of treatments acting on specific targets within the pathway to block/prevent lymphangiogenesis.
Current therapeutic options for lymphangiomatosis and Gorham's disease are limited. Given the rare nature of these disorders, there is a lack of clinical trials upon which to base treatment options. Case reports exist within the literature of the use of bisphosphonates, 17 interferon, 8 surgery, 6 and radiotherapy. 7 A recent study suggested that the anti-lymphangiogenic properties of rapamycin (sirolimus) and its derivatives may provide therapeutic value for the prevention and treatment of malignancies, respectively. 18 This knowledge has been translated from “bench to bedside” and sirolimus is now being trialed in patients with refractory lymphatic abnormalities. 19
Given our patient's extensive and progressive lymphangiomatosis with features of Gorham's disease (multiple vertebral body and pelvic bony involvement), we have considered multiple therapeutic options. Bisphosphonates were introduced relatively early in the disease course. They have not halted the lymphangiomatosis, but it is impossible to say whether they have slowed down the process. The patient continues to receive bisphosphonate infusion on a regular basis for the foreseeable future. Surgery has been largely unsuccessful given the extensive nature of the patient's disease. Radiotherapy was considered unsuitable for similar reasons. Interferon was not considered because of dose-related side effects. Lastly, we have commenced our patient on the mTOR inhibitor sirolimus in the hope that it halts the lymphatic proliferation within her skin, soft tissues, and bones. Her disease is so extensive that it significantly impairs her quality of life and is at risk of causing premature death. We await the results of treatment with interest.
Footnotes
Author Disclosure Statement
No competing financial interests exist.