
Abstract
Abstract
Lymphatic filariasis is the leading cause of secondary lymphedema wherein lymph transport is impaired due to lymphatic damage. FLT4 signaling and transcription factors such as FOXC2 play an important role in this type of lymphangiogenesis process induced by filarial parasites. The present study aims to assess the association of FLT4 and FOXC2 genes in lymphatic development/remodeling in lymphatic filariasis. A total of 118 lymphatic filariasis patients and 100 non-endemic and 50 endemic healthy subjects were enrolled for the present study. Allele-specific PCR and PCR-RFLP were adopted for the genotyping, and screening of FLT4 and FOXC2 genes was carried out by PCR-SSCP, followed by in-silico and statistical analysis. A novel variation (G357A SNP) was identified on FOXC2 gene screening that may have an effect on codon usage frequency during translational process. In FLT4, A3123G mutation was found in 3.39% of the case subjects but the functional role of this mutation, along with subject's clinical presentations and patient's age, emphasize its pathogenic role in lymphedema development. Two of the subjects exhibit compound heterozygosity (A3123G FLT4 mutation and G357A SNP of FOXC2 gene). As these two genes share a common pathway, we hypothesise a synergistic interaction of these two SNPs in inhibiting the downstream signaling resulting in lymphedema progression.
Introduction
T
Lymphatic filariasis is the leading cause of secondary lymphedema wherein lymph transport is impaired due to lymphatic damage. Among the 120 million people infected with the lymphatic dwelling nematodes, approximately 40 million have lymphedema and/or other pathologic manifestations, including hydroceles and episodic adenolymphangitis.2,3 The lymphedema characterized in filariasis is due to insufficient lymphatic drainage. The presence of adult worms triggers inflammation that may progress to fibrosis and subsequent obstruction of lymphatic vessels. 1 Adult filarial parasite within the lymphatic vessels of the host triggers a cascade of events that leads to abnormalities in lymphatic integrity and function.3,4 The dead and decalcifying adult worm elicit responses leading to lymphatic blockage and gross pathological lesions which, when coupled with pre-existing lymphatic dilation, may lead to damage of the lymphatic valves that may in turn induce lymphatic backflow and lymphedema. The aberrant lymph vessel development, innate immune responses that are triggered by the filarial antigen, ultimately result in the activation of vascular endothelial growth factors (VEGF), promoting lymph vessel hyperplasia as a first step to lymphedema development. Further the proinflammatory responses (IL-1, TNF-α), resulting from chronic lymphatic obstructions, render the lymphatic endothelial cells (LEC) hyperpermeable, leading to lymphedema. 5
Filarial parasites are capable of inducing lymphangiogenesis in vitro, a process that is LEC specific and relates to the excretory-secretory components of the filarial parasites and/or the elevated levels of circulating lymphangiogenic factors. 6 FLT4 signaling and transcription factors such as FOXC2 play an important role in this type of lymphagiogenesis process. Recent studies have implicated the role of VEGF gene family in filarial pathology.7–9 Furthermore, exposure to endosymbiont bacteria Wolbachia lipoproteins can cause innate and adaptive immune-mediated expression of VEGFs, promoting lymphangiogenesis, lymphatic endothelial proliferation, and dilation of lymphatic vessels precipitation in the development of chronic disease. 10
The FLT4 (fms like tyrosine kinase) gene is located at 5q35.3, whereas the FOXC2 (forkhead box C2) gene is located at position 16q24.1. Previous studies have implicated FOXC2 and FLT4 mutations in primary lymphedema.11–13 The current focus of filarial research is on finding the host genetic host determinants responsible for the varied clinical response in endemic populations.14–22 Therefore the present study aims to assess the role of FLT4 and FOXC2 genes in susceptibility for an increased risk in lymphatic development alterations and remodeling, especially in lymphatic filariasis.
Materials and Methods
The diagnosis of lymphatic filariasis in the present study was based on clinical evaluations and blood samples of 118 lymphatic filariasis cases referred by the National Filarial Control Program Center (NFCP), Siddipet, Andhra Pradesh, with the approval of the Ethical Committee. Pedigree information on the study subjects and their relatives were also obtained, and the families with at least one family member affected with lymphedema in addition to the proband were considered as familial aggregated cases. Microfilaria(Mf) positivity was checked based on microscopic examination of blood smears; circulating filarial antigen (CFA) detection was carried out by ICT test. Patients having any one of the clinical symptoms such as lymphedema, episodes of adenolymphangitis (ADL) or hydrocoel, and evaluated based on diagnostic criteria were included in the study. Individuals negative for Mf or CFA with absence of clinical symptoms were excluded from the study. Lymphedema grading was based on criteria described by Gyapong et al. 23 For a comparative analysis, 100 blood samples from age- and sex-matched controls obtained from non-endemic areas (Gandhi Hospital, Hyderabad) and 50 endemic donors from Siddipet, Medal district, were also considered.
DNA was extracted from blood samples following the rapid non-enzymatic method. 24 SSCP analysis for the screening of exon 18, 23, and 25 of FLT4 gene and FOXC2 whole exon was carried out. For convenience, the primer designing for FOXC2 was carried for six overlapping regions (F1–F6). Apart from SSCP, genotyping of −512C/T SNP of FOXC2 and A3123G mutation of FLT4 gene was also carried out by using PCR-RFLP and allele-specific PCR method, respectively. The primer sequences and PCR conditions are summarized in Table 1.
PCR assays were carried out in a 25 μL volume with 100 ng of genomic DNA, 10 pM of each primer, 2.0 mM dNTP (Merck, Germany), 1.5 mM MgCl2 and 10x PCR buffer [50 mM KCl, 500 mM Tris buffer, 160 mM (NH4)2SO4, pH 8.8, and 0.1% Tween 20], 0.1% Triton X-100, and 0.5U Taq polymerase (Sigma Aldrich). The thermal cycling was carried out in an Eppendorf Gradient Thermal cycler (Germany).
FOXC2 −512C/T SNP, screening was based on PCR-RFLP wherein the 306 bp PCR product was subjected to restriction digestion using 2U of Bbs I restriction enzyme at 37°C overnight. The samples were run on 2% agarose gel for genotyping of these polymorphisms. For FLT4 A3123G variation, allele-specific PCR was performed as described by Irrthum et al. 25
Comparison between lymphatic filariasis cases and the control group was performed by Fisher's exact test and Odds ratio. Disease association with individual SNP was analyzed by means of logistic regression to determine odds ratios (OR), and 95% confidence intervals (CI). 26 Difference between groups was statistically significant at p<0.05.
Results and Discussions
The demographic profile of the lymphatic filariasis cohort is presented in Table 2. The mean age at onset was 32.77±15.4 years. Females were predominant (66.1%) in the present cohort. The microfilaria rate was found to be 4.3%, whereas the antigenemia rate was 88.9%. Lymphedema was exhibited in 92.3% of the cases, of which 28.8% individuals had severe lymphedema (Grade III/IV) and 71.2% had a milder form of lymphedema pathology. 33.3% of the pedigrees exhibited a familial aggregation for the lymphedema. In individuals having lymphedema, 91.43% had a unilateral involvement of either lower or upper extremities or both. Only 8.57% of the cases showed bilateral involvement. Majority of the cases showed lower extremity lymphedema. With respect to the time since lymphedema onset, the mean duration is 7.79±11.9 years with 64.81% of the cases having lymphedema up to 5 years, while 14.82% and 12.96% of the cases suffered for 6–15 years and 16–25 years, respectively. Prolong duration (>25 years) was observed in only 7.41% of the cases. The prolong duration of the chronic disease is expected for a long-lived helminth such as W.bancrofti, with the mean expected lifespan being 8–15 years. 27
FOXC2 gene
SSCP analysis of FOXC2 F2 region revealed a band pattern variation in both lymphatic filariasis and control groups (Fig. 1). On sequencing a G>A transition at nucleotide position 357, a novel SNP resulting in a synonymous glutamine amino acid (Q117Q) residue at Codon 117 was identified (dbSNP rs199772307). The frequency distribution and the odds risk estimates of the FOXC2 A357G novel SNP and −512C/T SNP are given in Table 3.
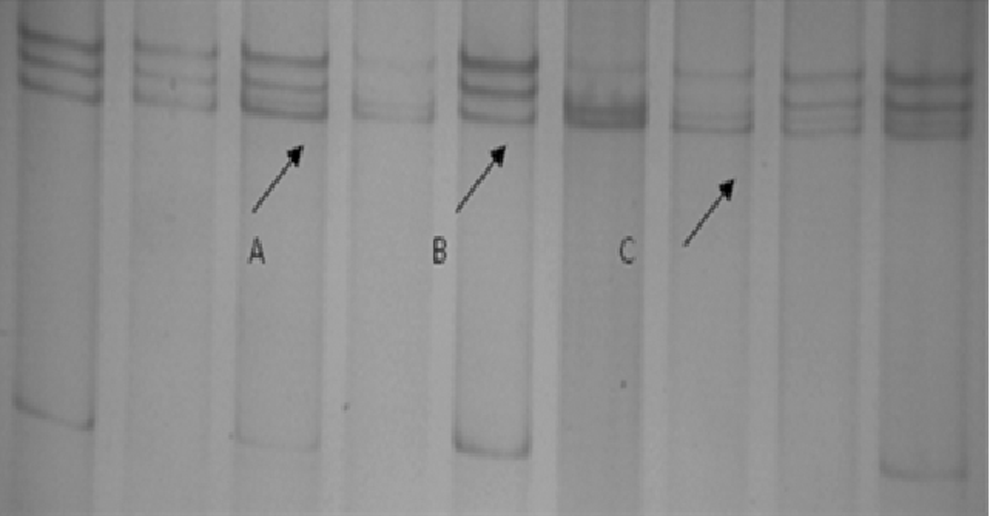
SSCP analysis of F2 region of FOXC2 gene.
The frequency of GG, GA, and AA genotypes were found to be 89.0%, 7.0%, and 4.0% in non-endemic; 83%, 10%, and 7.0% in endemic control, and 69.5%, 18.6%, and 11.8% in filariasis, respectively. The frequency of AA genotype was three-fold more frequent in lymphatic filariasis patients (11.8%) than in the non-endemic control subjects (4.0%). Similarly the AA genotypic frequency was 2.6 times higher in endemic controls than the non-endemic controls. As the variant allele of the SNP represented a high proportion in the cases, a possible functional role in the lymphedema can be speculated.
For odds risk estimates of G357A SNP, the comparison of the variant genotype AA with AG and GG for a risk predilection in the three inheritance models revealed a significant association of AA genotype with lymphatic filariasis with respect to non-endemic (AA vs. GA OR 3.80, CI- 1.20–12.0, p<0.001). Similarly there was a significant association of AA genotype with lymphatic filariasis when compared to endemic controls in recessive model (GA/AA vs. GG OR 2.70, CI-1.11–6.57, p=0.02). The A allele was also found to be significantly associated with lymphatic filariasis when compared to non-endemic (A vs. G OR 3.33, CI-1.75–6.45, p<0.001) and endemic controls (A vs. G OR 2.90, CI-1.35–6.38, p=0.02). Therefore the variant allele A is strongly associated with lymphatic filariasis. No significant difference in the genotype frequency distribution and odds risk estimates of FOXC2 −512C/T SNP was found in both the groups.
Distribution of the polymorphism based on gender, lymphedema grades, and familial aggregation is given in Table 4. The Odds risk estimates of the variant A allele did not show any significant variation with respect to familial aggregation (AA vs. GG OR-0.48 CI-0.16–1.44). However, lymphedema groups based on severity were examined for the possible association with the variant allele wherein a significant association with lymphatic filariasis was observed (AA vs. GG OR 2.81 CI-1.06–7.43, p=0.02). Therefore, A allele and its encoded product influence the severity of the lymphedema condition. In case of FOXC2 −512C/T, no significant difference in the odds risk estimates was found with respect to lymphedema severity and familial aggregation.
CI, Confidence interval; F, familial clustering cases; NF, Nonfamilial clustering cases; OR, Odds ratio.
Haplotype frequencies for the two SNPs of FOXC2 (−512C/T and G357A) were computed for their possible association with LF (Table 5). Frequency of wild-type CG haplotype was highest in both lymphatic filariasis and control group. The FOXC2 CA haplotype was found to be significantly associated with increased risk of lymphatic filariasis (CA vs CG OR 5.53 CI- 1.76–17.40, p=0.004) when compared to non-endemic controls, which indicates a synergistic role of the compound haplotypes in disease susceptibility.
In silico analysis of G357A novel SNP
Codon bias of synonymous codons is well established in different species, hence the difference in the frequency of the codon usage of FOXC2 G357A (CAG → CAA) synonymous SNP was also examined. Of all the four codons for glutamine, CAG is the most frequently used codon in Homo sapiens. Thus the G>A substitution results in replacement of a frequently used codon CAG (34.2) with a rarely used CAA codon (12.3). The codon usage analysis of FOXC2 cDNA shows that the rare codon CAA is used with a frequency of 0.04 in the gene and a codon change of CAG → CAA increases the frequency to 0.08. The other codons for Gln viz CAC and CAU are not in use. Earlier studies have shown that synonymous changes can significantly influence protein abundance via changes in translation efficiency. At the translational level, synonymous codon usage and codon-pair context (representing the pair of codons located in the A and P- ribosome sites) are under selective pressure since they affect mRNA decoding speed and accuracy. 28 Therefore, a replacement of a frequently used codon CAG with a rare one CAA can cause a ‘bottleneck effect’ in the translational rate and may affect the translational efficiency of FOXC2 mRNA.
The Unitprot and PDB domains structure predictions have depicted that the Q117 amino acid residue is located outside the forkhead domain and represents the helical strand of the protein chain. The synonymous SNP Q117Q lies in close proximity to the earlier reported mutations R121C, R121H and S125L in lymphedema-disctichiasis, affecting the forkhead domain.29,30 Interestingly, these missense mutations located outside of the forkhead domain still result in the lymphedema phenotype. R121C and S125L both may decrease the transcriptional activity, however the mechanism by which these mutations/genotypes influence is still unclear. Therefore, it is likely that the synonymous change influencing the codon usage and probably the translation efficiency may affect the forkhead domain folding during translational process and hence may have a regulatory role.
FLT4 gene
The SSCP analysis of exons 18, 23, and 25 coding for the tyrosine kinase II domain did not reveal any band pattern variation. However, the allele specific analysis of A3123G revealed the presence of G allele in four patients (3.39%), which when sequenced revealed a heterozygous mutation (Fig. 2).

Band pattern of A3123G genotypes of FLT4 gene.
The transition of A>G at nucleotide 3123 leads to a missense mutation with the substitution of histidine in place of arginine (CAC→CGC) in exon 23 at 1035 position. The missense mutation was previously reported in primary lymphedema patients. 25 Previous studies have shown that the substituted histidine is conserved in protein tyrosine kinases and in many serine/threonine kinases, suggesting a very important structural and/or functional role. 25 It is the first residue of the consensus sequence -HRDLAARN-, the central part of the catalytic loop of FLT4, wherein the aspartate residue is considered to be the key catalytic residue. 31 The histidine residue at position 1035 is located within the catalytic loop of tyrosine kinase II domain and hence may affect the downstream signal transduction pathway of FLT4 gene. 25 Earlier studies have reported that the lymphedema-associated mutant receptors were incapable of transphosphorylation in contrast to the normal receptor dimerization and intracellular tyrosyl transphosphorylation resulting from ligand binding during FLT4 activation.11,25 The H1035R mutation results in the loss of autophosphorylation capacity and hence loss of tyrosine kinase activity. The kinase inactive receptors degrade at slower rate than the wild-type receptors, and thus are more stable on the cell surface, which contributes to the development of lymphedema by reducing the relative amount of ligand binding to the wild-type FLT4 which leads to inefficient FLT4 signaling. 11 The kinase inactive FLT4 proteins were also poor activators of the downstream signaling cascades, suggesting that they fail to transduce VEGFC/VEGF-D signals into the endothelial cells. Animal model studies also have shown that the impaired signaling of membrane-bound FLT4 and soluble FLT4 due to inhibition of VEGFC results in complete destruction of the lymphatic network eliciting a lymphedema-like phenotype. 32
The clinical profile of the four patients harboring H1035R mutation is given in Table 5. The mutation was present in 3.39% of the patients in heterozygous state, and 75% of these patients had a history of chronic lymphedema. Interestingly, the pedigree of P2 showed a familial aggregation wherein lymphedema was reported in three generations. However, the DNA samples of the family members could not be obtained to substantiate the findings. Therefore, it may be hypothesized that this non-synonymous mutation A3123G is most likely to have a pathogenic impact on lymphedema development associated with lymphatic filariasis.
Interestingly, two of the case subjects having heterozygous FOXC2 G357A substitution were also heterozygous for the A3123 FLT4 mutation. The patients (P2 and P4) also had lymphedema symptoms as described in Table 6, clearly highlighting compound heterozygosity in the pathogenesis to lymphedema-associated lymphatic filariasis. FOXC2 is likely to be involved in determining the balance between lymph vessel growth promotion and inhibition. 33 During normal lymphatic development, maturing lymphatic vessels upregulate FOXC2, and VEGFC–VEGFR-2/3 signaling activates NFATc1, which together initiate a genetic program for formation of collecting vessels. Deficiency in either FOXC2 or active NFATc1 in the lymphatic endothelium affects the expression of target genes, shutting down the program for establishment of a collecting vessel identity and leading to a persistent capillary-like phenotype. 34 In vitro studies have shown that the mice heterozygous for both FOXC2 and FLT4 deficiency display similar lymphatic vascular defects as FOXC2-/- mice, suggesting cooperation between these two pathways and the FOXC2 acts downstream of FLT4. As FOXC2 is a downstream transcription factor in FLT4 signaling pathway, it is also possible that the G357A SNP of FOXC2 may have modifying effect that results in the mitigation of the main effect of the other known disease causing SNP/mutation within the FOXC2 gene or in the other signal pathway genes (including FLT4) affecting the lymphangiogenesis process. Therefore the synergistic heterozygosity of FOXC2 G357A SNP and FLT4 A3123G SNP may have a possible role in lymphedema development.
Lymphatic endothelial cells (LECs) actively participate in the phenotypic consequences of a disordered lymphangiogenesis relating to tissue fluid accumulation in the pathogenesis of lymphedema. Hence, understanding the interaction between lymphatic endothelial cells and the filarial parasites may provide insights into the lymphedema pathology. In the present study, a novel variation (G357A SNP) was identified on FOXC2 gene screening which, based on in silico analysis, may have an effect on codon usage frequency during translational process. The FLT4 A3123G mutation was found in only 3.39% of the case subjects but the functional role of this mutation along with subject's clinical presentations and age emphasize its pathogenic role in lymphedema development related to lymphatic filariasis, but large population-based studies and functional analysis of this mutation can establish the role of this mutation as a putative cause of secondary lympehdema. As two of the subjects exhibit heterozygous A3123G FLT4 mutation and G357A SNP of FOXC2 gene, it may be speculated that the compound heterozygosity may lead to synergistic interaction of these two SNPs in inhibiting the downstream signaling, resulting in lymphedema progression. Since the current studies are focused on targeting genetic basis of pathogenesis, our findings contribute to the putative targets in which the FLT4 gene mutation was found to have a significant role in lymphatic filariasis, while FOXC2 and FLT4 SNP interactions need to be further elucidated at a functional level.
Footnotes
Acknowledgments
We thank all the patients who participated in this study; YS is a recipient of Senior Research Fellowship from University Grants Commission (UGC), SFQ is a recipient of Senior Research Fellowship from University Grants Commission (UGC) under the MANF scheme, and the study is funded by Indian Council of Medical Research (ICMR), New Delhi India (2009–2012).
Author Disclosure Statement
The authors declare that they have no competing financial interests.