
Abstract
Abstract
Background:
Stewart-Treves syndrome is a rare complication of breast cancer treatment, representing a lymphangiosarcoma commonly associated with lymphedema and severely impacting patient's outcome. The tumor typically develops in the atrophic, pachydermatous, hyperkeratotic skin of limbs affected by long-standing lymphedema. Clinical data associated with Stewart-Treves syndrome and lymphedema management have rarely been published.
Methods and Results:
In the period between 1980 and 2009, ten patients with Stewart-Treves syndrome were diagnosed and treated at the Foeldiklinik, Hinterzarten, Germany. Nine of the ten patients were female. Five patients had previously suffered from breast cancer (and were treated with mastectomy); two from other malignancies; two patients had primary lymphedema, and one had undergone lower extremity lymphadenectomy. All cancer patients had undergone radiation treatment. In all cases, the sarcoma developed in non-irradiated areas 6–48 years (average 16.3 years) after the onset of lymphedema. None of the patients had received complex decongestive physical therapy (CDT). Two patients had above-elbow amputation, one had shoulder exarticulation, two patients had wide excision and skin grafting, two patients had above-knee amputation procedure, two patients had a below-knee amputation procedure, and one patient had no surgical treatment at all. The time to recurrence after surgery, time to metastasis, patient survival and CDT were recorded.
Conclusions:
Patients with lymphedema should be closely examined starting 5 years from the time of lymphedema onset, paying special attention to those with associated malignancies. Only early diagnosis and treatment by radical ablative surgery confers a reasonable prognosis with this rare but aggressive disease. A potential effect of CDT on lymphangiosarcoma has to be studied in a greater patient cohort.
Introduction
F
Lymphangiosarcoma has been described in Milroy's disease, in idiopathic, congenital, traumatic, and filarial lymphedema.7–11 This tumor typically develops in the atrophic, pachydermatous, hyperkeratotic skin of limbs affected by long-standing lymphedema and presents as a purplish patch that then develops into a plaque or nodule (Fig. 1), around which small satellite areas develop and become confluent, forming an expanding lesion. It may ulcerate, leading to episodes of bleeding and infection. Necrosis may be evident in advanced stages.12,13
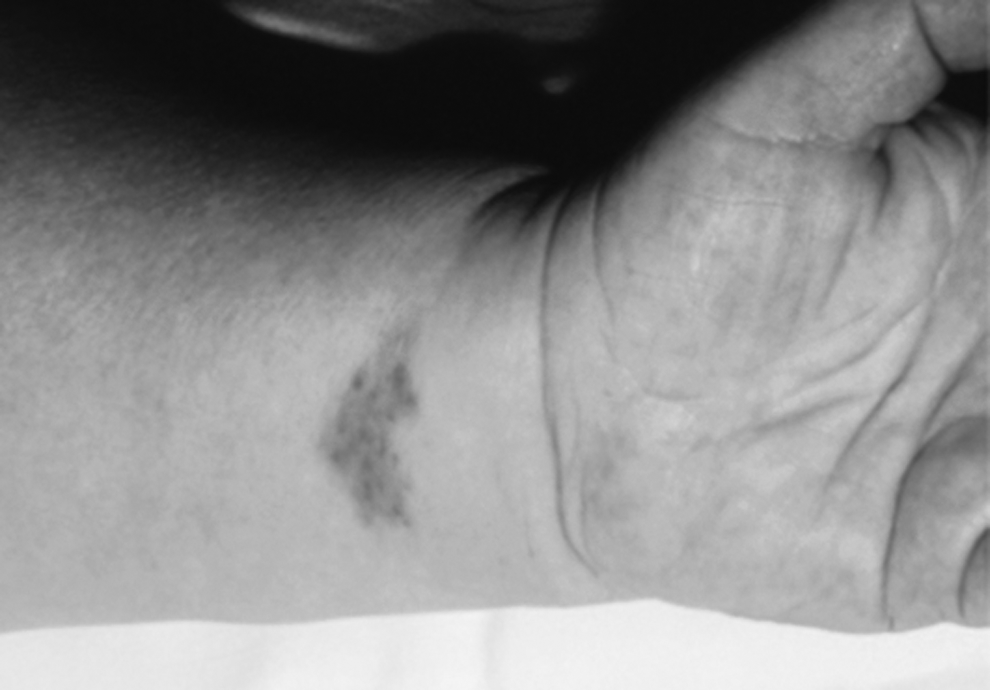
Clinical presentation of lymphangiosarcoma as a purple patch resembling a benign ecchymotic lesion.
Reports describing the magnetic resonance (MR) imaging features in patients with Stewart-Treves syndrome are rare.14–17 The involved lymphedema is detected on T2-weighted or turbo inversion recovery magnitude (TIRM) sequences by high signal intensities in the subcutaneous region with a honeycomb pattern (Fig. 2a). The actual tumor itself demonstrates variable signal intensities on T2- or TIRM-weighted images, low signal intensities on native T1-weighted images, and high signal intensities on T1-fat-saturated images after intravenous contrast injection (Fig. 2b). By performing MR-examination in patients with a suspected lymphangiosarcoma, it is possible to detect and determine its infiltration into the surrounding structures precisely. This examination therefore yields important information to plan biopsy or surgical treatment. Histologically, lymphangiosarcoma is a proliferation of malignant endothelial cells with epitheloid morphology, hardly recognizable vascular channels, but immunohistological features of lymphatic endothelium (Fig. 3).4,18

Forty-three year old female with chronic secondary lymphedema of the left lower leg after inguinal lymph node extirpation.
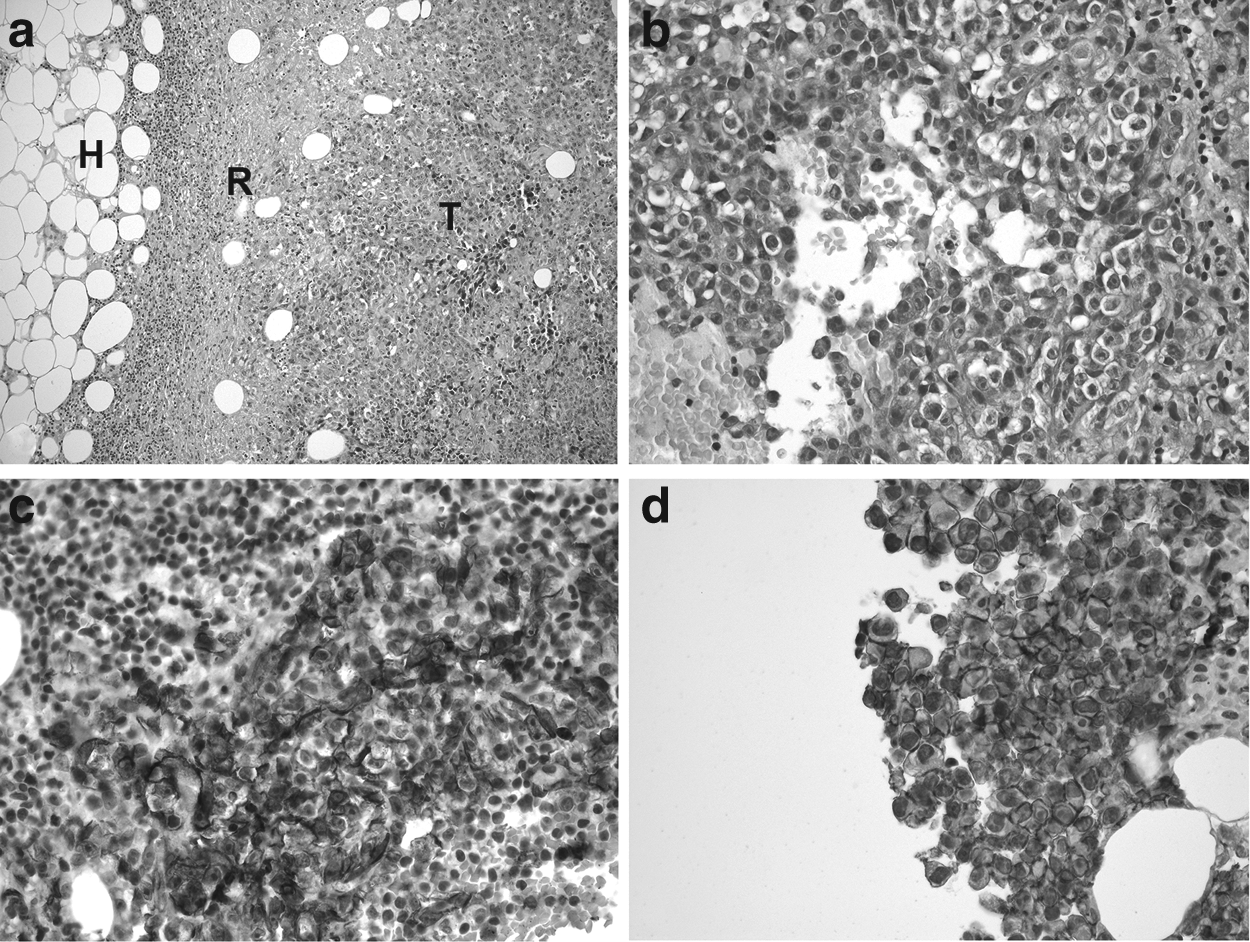
Histological features of G3 lymphangiosarcoma in Stuart-Treves syndrome.
In this study, we review 10 cases of lymphangiosarcoma at the Foeldiklink, Specialist Center for Lymphology Hinterzarten, Germany, from 1980 to 2009, with a specific interest in its association with conservative lymphedema management.
Material and Methods
Patients
Ten cases of histologically confirmed lymphangiosarcoma at the specialized clinic for lymphology, Foeldiklinik Hinterzarten, between January 1, 1980, and January 1, 2009 were reviewed. The underlying condition leading to lymphedema and the latency from the time of diagnosis of lymphedema to the diagnosis of lymphangiosarcoma was noted. The time to recurrence after surgery, and patient survival were recorded. Particular attention was paid to the fact whether the patients had received complex decongestive physical therapy (CDT). 19 The procedures presented here are in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008.
Histology and immunohistochemistry
The tumor biopsy was taken from non-irradiated areas of the limbs through normal, biopsy-taking procedure. The tissue was formalin-fixed, paraffin-embedded and cut in 4 μm slices. The slices were stained with hematoxylin and eosin (HE). Immunohistochemistry was performed according to standard protocols with the antibodies (all ready to use, Dako Cytomation, Glastrop, Denmark) to detect the following antigens: CD31 (PECAM 1) as a specific pan-endothelial marker; D2-40 (Podoplanin) as a specific marker for lymphatic endothelial cells; CD34, a specific blood endothelial marker, Ki-67, a proliferation marker), and the proto-oncogene FLI-1 (dilution 1:50, Santa Cruz Biotechnologie Inc., Heidelberg, Germany). The Envision Flex™ System (Dako Cytomation, Glastrop, Denmark) was used as a detection system.
Therapy
The surgical treatment of the lymphangiosarcoma was classified into three categories as i) wide excision and skin grafting, ii) above the elbow amputation, above the knee amputation (AKA), and iii) below the knee amputation (BKA).
Results
Among the more than 15,000 patients treated at the Foeldiklinik during 1980 and 2009, ten cases of lymphangiosarcoma were retrieved. Nine of the ten patients were female. Five patients had previously suffered from breast cancer; two patients had other malignant conditions; two patients had primary lymphedema, and one had undergone lower extremity lymphadenectomy. All but the latter three patients had undergone radiation treatment for their primary condition. In all cases, the sarcoma developed in non-irradiated areas. The main clinical features of these patients are shown in Table 1, in which the patients are listed in chronological order of presentation.
The diagnosis of lymphangiosarcoma was made 6 to 48 years (average 16.3 years) after the onset of lymphedema. In the patients with post-irradiation lymphedema, the lymphangiosarcoma appeared as early as 6 years after onset of lymphedema (average 9.1 years). Patients without primary malignancy (and consequently without history of radiation treatment) demonstrated a much longer latency, averaging 33 years, between the onset of lymphedema and the diagnosis of lymphangiosarcoma.
Table 1 demonstrates patient survival, and shows that only one of the breast cancer patients survived as long as 36 months after surgery, while two of the primary lymphedema patients survived 36 or 47 months. However, none of the patients in our series received CDT. Shoulder exarticulation was performed in one case, high arm amputation in another, above-the-knee-amputation in three cases, below-knee-amputation in two cases, and wide excision and skin grafting in two cases. One patient did not receive surgical treatment in the context of metastatic disease.
Discussion
The single most important causative agent in Stewart-Treves syndrome is prolonged chronic lymphedema. Although Stewart-Treves syndrome develops after radical mastectomy in most patients, lymphangiosarcoma also develops in other forms of primary and secondary lymphedema. Edema secondary to cardiac or renal disease is not associated with this malignancy. Thus, edema alone is not sufficient to cause lymphangiosarcoma. Therefore, additional factors such as a genetic predisposition may also be required.
The precise pathomechanisms of lymphangiosarcoma have remained controversial. Causative factors such as local immunodeficiency 20 and radiation,21–23 and the association of the disease with hypertension and cardiovascular disease 24 have been described. In their original series, Stewart and Treves 2 reported that the earliest angiosarcoma appeared 6 years after radical mastectomy, the longest interval being 24 years (average 12.5 years). In 1981, Yap et al. reported a series of 22 patients, in whom the median interval from mastectomy to the onset of angiosarcoma was 11 years (range, 5–16 years). 27
Our data confirm previous reports that primary lymphedema patients have a longer latency.25,26 In our series, one of the ten patients survived as long as 3 years, having undergone an above-knee amputation. However, the type of operation does not seem to affect the prognosis, providing that the margins are histopathologically tumor free. Since its description by Stewart and Treves in 1948, case series and reports have repeatedly confirmed the fatal character of this aggressive condition, which we are still far from understanding. Despite aggressive and multimodal treatment, survival beyond 24 months remains the exception.27,28 Therefore, it is appropriate to stress the importance of prevention and early detection of the disease.
A conservative approach is undesirable even in clinically nonmetastasized lymphangiosarcoma. Radical ablative surgery has been advocated by many as the approach of choice, providing the best chance of long-term survival.29,30 Chemotherapy,31–34 immunotherapy, 35 and radiation therapy32,36,37 have been reported as stand-alone treatment or as adjuvants to surgery. Despite multimodal and aggressive management, lymphangiosarcoma is associated with a high rate of local recurrence and metastasis. 38 As there is a 100% correlation between the development of lymphangiosarcoma and the presence of clinically manifest lymphedema, the question should be asked whether the incidence of the disease is significantly altered by adequate lymphedema treatment, or the prevention of lymphedema.39–43
In our series, none of the patients affected by lymphangiosarcoma had received CDT, neither after mastectomy nor after radiation therapy. The reasons for this may be manifold. Of note, CDT has been shown to be the most effective therapy in reducing lymphedema and improving quality of life44–49 and superior to physical therapy alone. 50 At the molecular level, reduced expression of pro-inflammatory genes has been demonstrated after CDT, suggesting that inflammation and tumor progression may be linked in lymphedema. 51
In conclusion, we recommend that patients with lymphedema should be examined at close intervals starting 5 years from the time of lymphedema onset, paying special attention to those with malignant lymphedema. Lesions with high similarity to hematomas, and indurations in the subcutaneous plane, should be diagnosed with great care.
Footnotes
Acknowledgments
We thank J. Wilting and K.P. Aung for their critical review of the manuscript.
Author Disclosure Statement
The authors declare no competing financial interests.