
Abstract
Abstract
Background:
Lymphatic endothelial cells (LECs) derived from lymphatic malformations (LMs) bear activated PIK3CA alleles yet display an inflammatory gene expression profile. A basis for the inflammatory phenotype was sought by screening for coexisting somatic mutations.
Methods
Conclusions:
Inflammatory stress displayed by LM-LECs is consistent with errors in lipid metabolism that may be linked to acquired mutations. The acquisition of PIK3CA alleles may be a permissive event that antagonizes inflammation and metabolic defect.
Introduction
L
PIK3CA is the catalytic subunit of the class 1A lipid phosphorylase, phosphoinositide 3-kinase (PI3Kα). 11 PI3K links cell-surface receptors for ligands such as growth factors [vascular endothelial growth factor (VEGF)], hormones (insulin, for example), and inflammatory stimuli to cellular effectors that modulate cell growth, metabolism, and survival. Upon receptor activation, PIK3CA is tethered to the receptor complex by the regulatory subunit of PI3K, p50–55/p85. This conditional activation of PI3K results in the phosphorylation of substrate phosphatidylinositol 4,5-bisphosphate, producing the second messenger phosphatidylinositol 3,4,5-trisphosphate. Normal regulation can be circumvented by the constitutive activation of PIK3CA through mutation, a common occurrence in a variety of cancers.12,13 PIK3CA activating mutations occur predominantly in two “hotspot” regions of molecule: the helical domain that interacts with p85α (residues 542 and 545) and in the kinase domain (at residue 1047). Activated PIK3CA has been associated with tissue hyperplasia.13,14 When targeted to endothelial cells in animal models, the H1047R mutation resulted in vascular hyperproliferation and lethality. 15 Thus, activating PIK3CA alleles, acquired somatically by LECs, are likely to enhance proliferation, promoting vessel expansion that is characteristic of LM. It is unclear if other genetic factors contribute to LM.
Previously, we showed that LM cell populations display a gene expression profile indicative of cellular activation or inflammation. Isolated LECs from LM overexpressed VEGFC and COX-2. In this report, we elaborate further on the shared inflammatory characteristics of LM-LEC populations. We used chemical inhibition and protein immunoblotting to probe the signaling pathways involved in the inflammatory process. We also sought evidence of mutations other than PIK3CA alleles that might drive LM development. Our findings implicate gene-based errors in metabolism as an underlying cause of cellular inflammation. Inflammation, largely independent of PI3K activity, was regulated, in part, by AMPK. AMPK is a regulator of cell growth, inflammation, and metabolism. 16 PI3K action antagonizes that of AMPK, 17 and its constitutive activation may be a permissive adaptation that counters the negative impact of metabolic errors. Expanding our understanding of the genetic and metabolic variables that contribute to LM will benefit the identification of novel targets for future LM treatments.
Materials and Methods
Materials
Primary cells derived from patient tissue or Lonza Corp (dLyNeo, dermal LECs) were propagated in EGM-2MV (endothelial cells) or DMEM (nonendothelial cells) obtained from Lonza. Cellular nucleic acid was prepared using extraction kits from Invitrogen (PureLink Genomic DNA Mini Kit) and QIAGEN (RNeasy Plus Mini Kit). DNA fragments were cloned (Zero Blunt PCR Cloning Kit; Life Tech) into pDONR221 and sequenced from M13/pUC reverse sequencing primer supplied by Thermo Fisher Scientific. cDNA was prepared with High Capacity cDNA Reverse Transcription Kit from Applied Biosystems. Amplification was performed with SYBR premix Ex Taq II (Takara). Protein lysates were prepared in RIPA lysis buffer (Santa Cruz Biotech.). Antibodies obtained from Cell Signaling Technologies include: anti-AKT/phospho-AKT (#4691/#4060); anti-AMPK/phospho-AMPK (#5832/#2535); anti-p42/44 (ERK1/2) and anti-phospho-p42/44 (#9102 and #4370); anti-COX-2 (#12282); and anti-HO-1 (#5853). Mouse anti-human GAPDH was obtained from Fitzgerald (10R-G109A). Rabbit anti-podoplanin was purchased from Sigma-Aldrich. Anti-human CD31 and anti-rabbit IgG magnetic beads were supplied by Miltenyi. HRP-conjugated anti-rabbit and anti-mouse antibodies and Clarity ECL substrate were purchased from Bio-Rad. Restore PLUS was from Thermo Fisher. WST-1 reagent was supplied by Clontech. Additional chemicals included chloroquine, BSA, palmitic acid, and thapsigargin from Sigma-Aldrich and GDC-0941, dorsomorphin (compound C), AICAR, rapamycin (sirolimus), and etomoxir from Selleck Chemicals.
Cell isolation and culture
Access to patient tissue or body fluids and the isolation of LECs have been described elsewhere. 3 Access and experimental handling of patient samples was conducted with CCHMC Institutional Review Board approval and in accordance with national guidelines. The clinical identification of lesions was made by the attending physician in collaboration with members of the CCHMC Pathology Department. LECs derived from LM were selected by CD31 and podoplanin expression. Cells were expanded in EGM-2MV. Nonendothelial cells harvested from lesions were grown in DMEM containing 10% fetal bovine serum (FBS). Gene expression analyses were conducted on endothelial cells cultured for 24 hours in reduced EGM-2MV medium, which lacked growth factors and contained 0.5% FBS.
Genomic sequence analysis
Genomic DNA was isolated from LM-LECs, nonendothelial lesion-derived cells, or cells harvested from patient blood. Somatic mutations were identified by whole exome sequencing (WES) comparing LM-LEC sequences to sequences from either homologous blood or lesion non-ECs. Verification of identified mutations was accomplished by direct Sanger sequencing of polymerase chain reaction (PCR)-amplified DNA fragments spanning the target base or Sanger sequencing of DNA fragments cloned into sequencing vectors. Sanger sequencing was performed by the CCHMC DNA Sequencing Core. The identification of major PIK3CA alleles was accomplished by ARMS-PCR as previously described. 3
Whole exome sequencing
Exome library preparation was performed through the Cincinnati Children's Genomic Variation and Gene Discovery Core using NimbleGen EZ (Roche, Basel, Switzerland) and sequenced using the Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA), according to manufacturer protocols. Genomic alignment was performed using the Broad Institute Integrative Genomic Viewer 18 with modifications according to their “Best Practices” guideline.
Variant filtering
Variants were evaluated with a read depth of >10 and quality index >60. Given the comparison-based method, these lower stringencies were expected to increase the sensitivity of detection without significantly increasing the number of variants for analysis. Intronic and synonymous coding variants were excluded from analysis.
Variants were considered in two models: somatic de novo or loss of heterozygosity (LOH). In the former case, variants were only retained if they were present in the lesion tissue, but absent in the control tissue. Only novel variants were analyzed under the assumption that an activating mutation would result in embryonic lethality. For LOH, variants were retained if they were present as heterozygous in the control tissue, but homozygous in the lesion. Remaining variants were further analyzed for the possibility of alignment errors. The variants were compared to other samples from the same run. If the variant was found in a normal tissue type from another sample, it was discarded.
Variant analysis
Potential pathogenicity of a candidate mutation was assessed by evaluating the strength of the mutation through sorting intolerant from tolerant (SIFT) (sift.jcvi.org), PolyPhen (software.embl-em.de/software/8), and MutationTaster (mutationtaster.org).
Gene expression analysis
Transcriptional gene expression was measured by quantitative RT-PCR (qRT-PCR) on cells grown in reduced EGM-2MV for 24 (prescreens) or 6 hours (quantitative assays). PCR was performed on diluted cDNA (1:20) in a CFX Connect real-time thermocycler (Bio-Rad).
Primers used in this study (5′ to 3′; forward followed by reverse): [VEGFC] CACGAGCTACCTCAGCAAGA and GCTGCCTGACACTGTGGTA; [COX-2] TCATCTGCAATAACGTGAAGGGC and GGAGCGGGAAGAACTTGCATT; [HO-1] CGGGCCAGCAACAAAGTG and AGTGTAAGGACCCATCGGAGAA; [IL-8] TCTGCAGCTCTGTGTGAAGG and ACTTCTCCACAACCCTCTGC; [ESEL] GGTTTGGTGAGGTGTGCTC and TGATCTTTCCCGGAACTGC; [ANGPTL4] TCTCCGTACCCTTCTCCACT and AGTACTGGCCGTTGAGGTTG; [CHOP] AGAACCAGGAAACGGAAACAGA and TCTCCTTCATGCGCTGCTTT; and [sXBP1] CTGAGTCCGAATCAGGTGCAG and ATCCATGGGGAGATGTTCTGG.
Assays were performed in triplicate on two (prescreening) or three independent samples. Cell cultures were treated with PI3K inhibitor GDC-0941 at a concentration of 1 μM or AMPK inhibitor Dorsomorphin at 2 μM. Protein immunoblotting was performed with cell lysates prepared in RIPA lysis buffer, fractionated in 4%–15% polyacrylamide gels. Protein was blotted onto nitrocellulose and treated sequentially with primary rabbit antibodies, HRP-linked secondary antibodies, and enhanced chemiluminescence solution. Visualization was by digital imaging (ChemiDoc MP Imager; Bio-Rad).
Induction of metabolic stress
Logarithmic cultures in EGM-2MV were treated with Chloroquine (CQ) at 40 μM, etomoxir (Etom) at 200 μM, BSA-conjugated palmitic acid (cPA) at 40 μM, or thapsigargin (Thap) at 20 nM. Serum starvation was accomplished by washing monolayers with warm starvation medium (0.1% FBS) and replacing with same. High serum (HS) exposure was performed in medium containing 20% FBS. Cultures were harvested for RNA after 24 hours of culture.
Drug inhibition studies
The sensitivity of LM-LECs to growth inhibition by rapamycin was determined as previously described. 3 In brief, cells were plated at 1000–2000 cells per well of a 96-well dish, and rapamycin was added after 24 hours. Proliferation was measured after a further 48-hour incubation by reaction with WST-1 Reagent. Three independent assays were performed in triplicate wells. Similarly, cells were treated with AICAR over a concentration range of 1 mM (from a 25 mM stock in PBS) to 16 μM (twofold dilutions). Proliferation is expressed as the percent of maximum growth (untreated cells) above cytostasis levels (cells treated with 50 nM rapamycin).
Statistical evaluations
Statistical treatments and graphing were carried out using GraphPad software (Prism7). Error bars denote standard deviations, and levels of significance were determined from simple t tests. Linear regression curves were generated by Prism7.
Results
Source and derivation of LM-derived LECs
Background information on patient and lesion samples is provided in Table 1. Fourteen independent LM samples were studied, of which five were included in our previous report. 3 Three samples were from patients diagnosed with PIK3CA-related overgrowth syndrome (PROS). Remaining samples came from tissue or fluid of surgically reduced localized LM. Patient access was undertaken in accordance with institutional guidelines (IRB approval) and with patient consent. LECs were isolated following CD31+ and podoplanin-positive selection. During this study, three LM-LEC isolates (1381, 1565, 1505) became increasingly difficult to propagate preventing their inclusion in all analyses. These cultures contained an increasing fraction of enlarged multinucleated cells suggestive of cellular senescence. Each cell population in the study harbored one of five predominant activating PIK3CA alleles 10 as determined by ARMS-PCR. 3
LM-LEC isolates are listed as numerical patient de-identifiers. The harbored PIK3CA allele is shown along with the original clinical diagnosis. The form of the patient sample (aspirated fluid or surgical tissue specimen), anatomical site, and scope of involvement (diffuse or focal) are presented.
CLOVES, complex lipomatous overgrowth, vascular malformation, epidermal nevi, and skeletal anomalies; CVLM, capillary-venous lymphatic malformation; LEC, lymphatic endothelial cell; LM, lymphatic malformation; KTS, Klippel–Trenaunay syndrome; VLM, veno-lymphatic malformation.
Exome sequencing of LM-LECs reveals coexisting gene variants
WES of six patient samples was undertaken to reveal non-PIK3CA gene variants that may contribute to LM. Somatically acquired mutant alleles discovered by these analyses are listed in Table 2. Single candidate variants were found in five of the six samples. Read frequencies for each approximated those associated with the coharbored PIK3CA allele. Variants present in LM-LECs were confirmed by direct sequencing of genomic DNA or cloned sequences (isolate 935) derived from genomic DNA (Fig. 1). Variants were single-base substitutions in coding regions of the respective genes. In sample 935, a novel variant of the autophagy-related protein ATG2A was present. ATG2A is physically associated with lipid droplets and is reported to regulate their metabolism.19,20
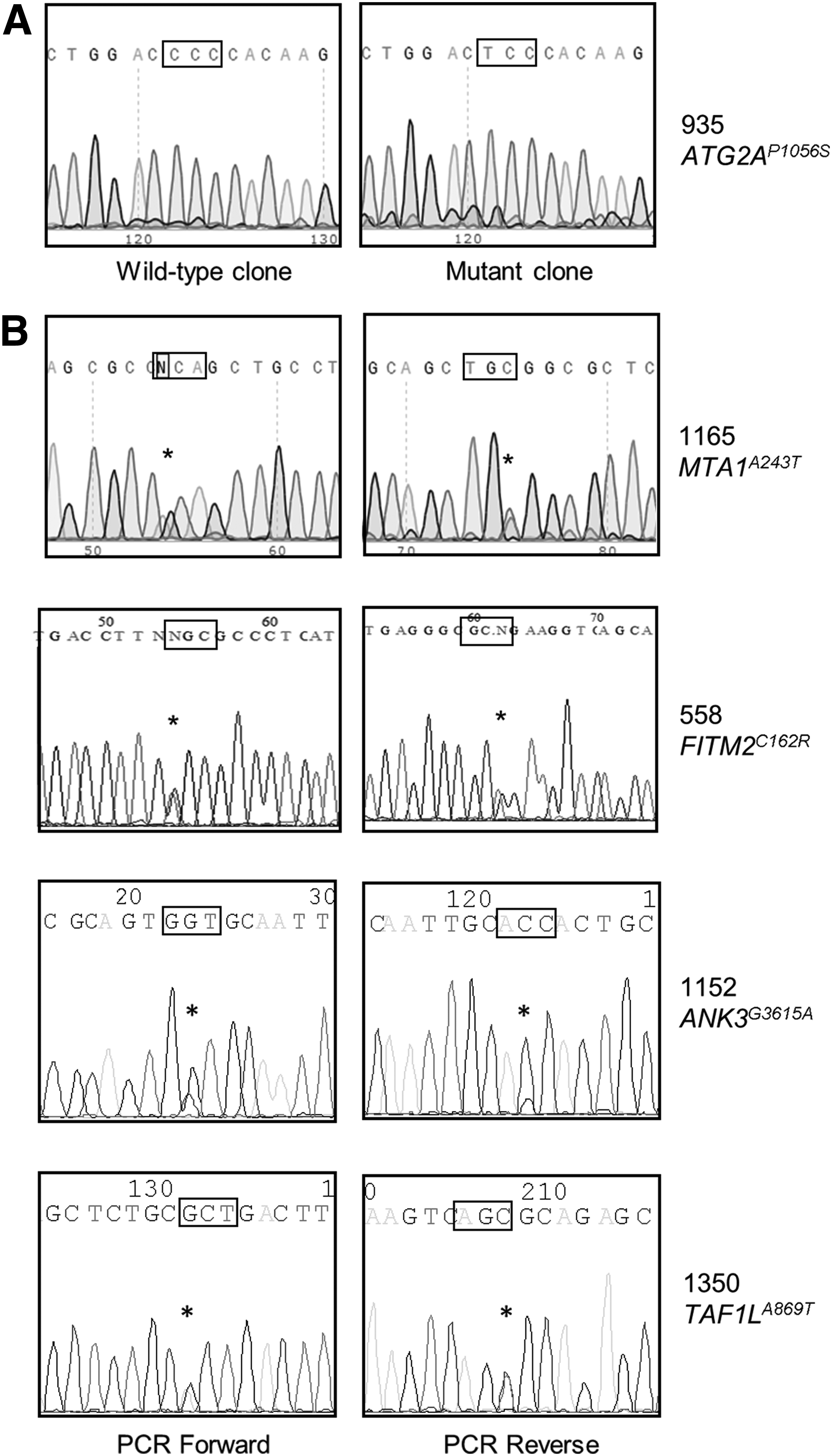
Verification of mutant alleles by Sanger sequencing.
Gene variants discovered in LM-LEC populations by WES are shown. Variant candidacy was determined based on read depth, read quality, and likelihood of functional defect. Base positions are shown (UCSC genome browser hg38 assembly), with base substitution, amino acid change with residue position, and the affected gene. Allele frequencies represent read frequencies for the respective mutations.
WES, whole exome sequencing.
A variant of MTA1 (metastasis-associated protein 1) was discovered in sample 1165. MTA1 is a component of the NuRD (nucleosome remodeling and deacetylation) complex that functions in regulating gene expression. 21 Sample 558, from a patient with CLOVES, carried a novel allele of FITM2. FITM2 (fat storage-inducing transmembrane protein) participates in the formation of lipid droplets. 22 A variant of ANK3 was identified in 1152. The ANK3 gene gives rise to multiple, differentially spliced protein products. 23 The affected base in the 1152-related allele appears in small protein products of ANK3. Their function is unclear although reports suggest involvement with endosomal- or lysosomal-directed transport.24,25 Finally, a variant of TAF1L was discovered in sample 1350. TAF1L is a gene regulator of unknown function. 26 No qualifying gene variant, apart from PIK3CA, was identified in the sixth sample from patient 837.
PI3K-independent inflammatory responses of LM-LECs
Previously, we showed significantly higher levels of COX-2 and VEGFC expression in five populations of LM-LECs relative to control LECs (dLyNeo). We extended this analysis to include nine more recently derived populations and additional inflammatory gene products. In Figure 2, we show the results of a qRT-PCR screen which highlight the variable upregulation of COX-2, heme oxygenase-1 (HO-1), IL-8, e-selectin (E-SEL), and VEGFC among the LM-LEC cultures. The LM-LEC cultures were ordered from left to right in the different panels based roughly on relative COX-2 expression.
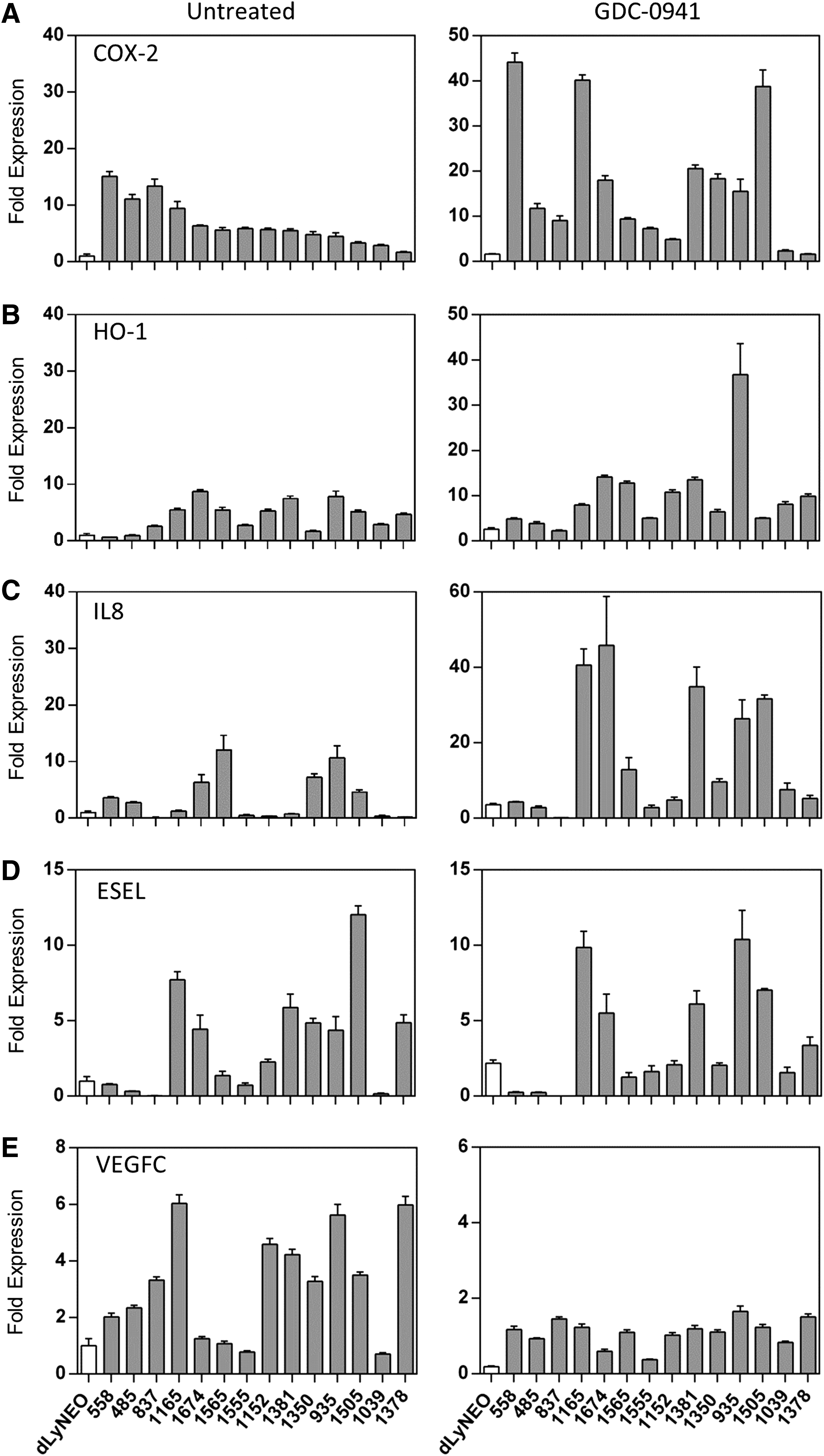
PI3K-independent expression of inflammatory genes in LM-LEC. The expression of inflammation related genes
The three LM-LEC samples on the extreme left (558, 485, 837) were derived from patients with clinically diagnosed PROS. The role of activated PI3K on inflammatory gene expression was examined by treating cells with the potent PI3K inhibitor, GDC-0941 (Fig. 2, right panels). The expression of VEGFC, which was generally upregulated in LM-LECs, was reduced to a common level upon inhibition of PI3K. In contrast, expression of COX-2, HO-1, and IL-8 was unaffected or variably increased upon PI3K inhibition. E-SEL expression appeared little affected by PI3K inhibition.
The relationship between inflammatory gene expression and PI3K activity was examined further by correlating cellular levels of AKT phosphorylation (as a measure of PI3K activity) to the expression of COX-2 and HO-1 protein in individual LM-LEC samples (Fig. 3). This was relevant because acquired PIK3CA alleles conferred measurably different levels of PI3K activity. The H1047L/R and E545K allele-bearing cells displayed a higher constitutive level of AKT phosphorylation than did E542K and E545A allele-bearing cells (11.5 ± 0.7-fold vs. 6.5 ± 0.3-fold elevation over control LECs; p < 0.001) (Fig. 3A, B). COX-2 protein levels were elevated in LM-LEC populations with statistical significance attained in 7 of 12 cultures (Fig. 3C). Higher COX-2 expression was associated with alleles H1047/E545K (Fig. 3C, right panel; p = 0.044).
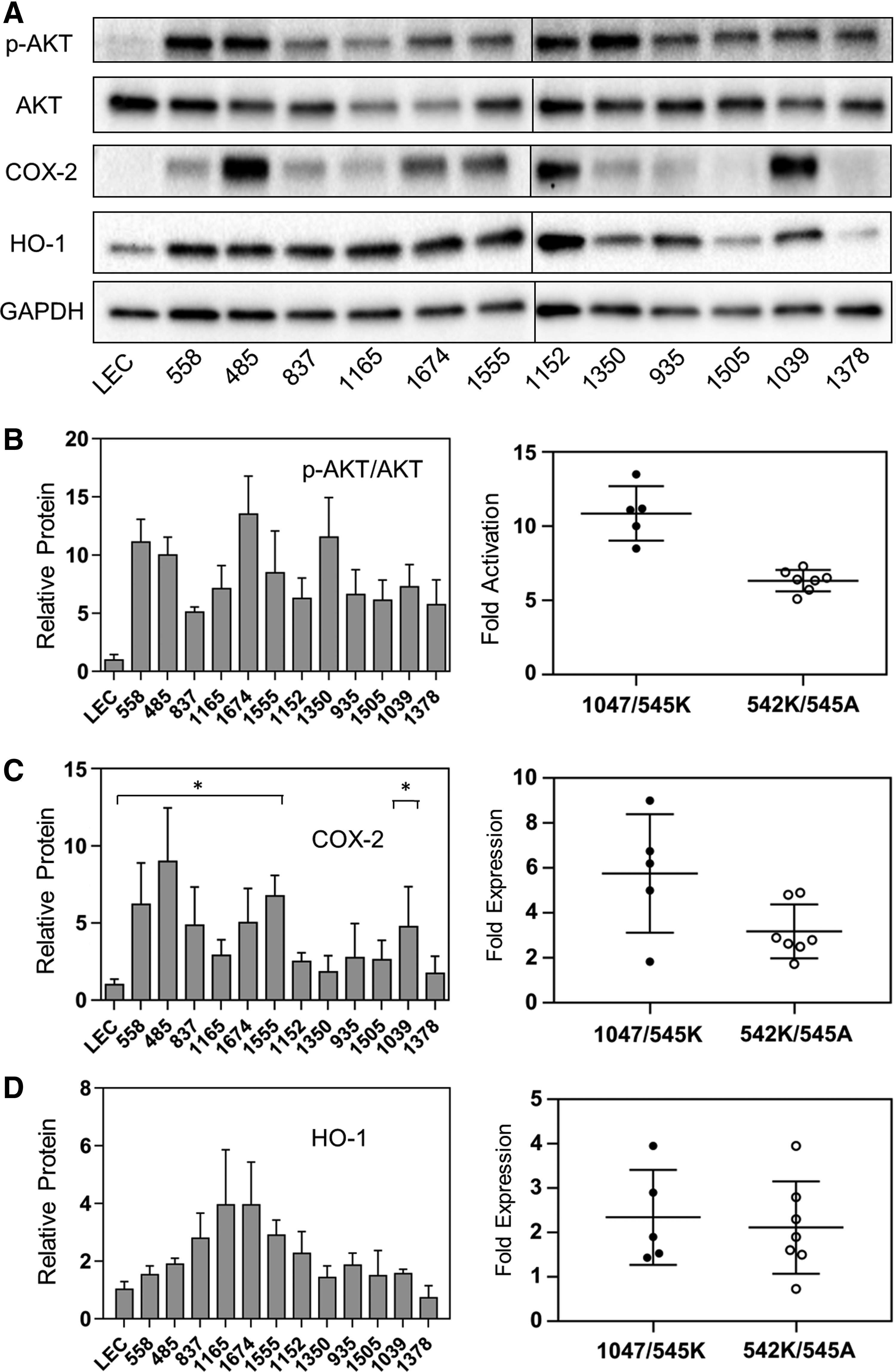
PI3K activation and inflammatory protein expression. Immunoblots of LM-LEC isolates (a representative blot, of three independent experiments, is shown in
HO-1 protein was also elevated in LM-LEC with statistical significance achieved in 6 of the 12 cultures (Fig. 3A, D). Levels of HO-1 expression lacked any correlation with PI3K activity (right panel, Fig. 3D).
AMPK regulates the inflammatory gene expression
Evidence of PI3K-independent inflammatory activation and the presence of mutations that potentially impacted cell metabolism raised the possibility that cells were metabolically stressed. Lipid-related stress, suggested by the presence of gene variants that could impact lipid metabolism, provided a rationale for examining the activation of AMPK in LM-LECs. AMPK is a stress and energy sensor that modulates inflammatory activation and activates lipid mobilization at the level of lipid droplets.27,28 Levels of AMPK activity were determined from immunoblots of phosphorylated AMPK in cell extracts (Fig. 4A). Levels in LM-LECs were statistically indistinguishable from control LECs (Fig. 4A, B) despite the constitutive activation of PI3K and its reported negative effect on AMPK activation.17,29 We treated LM-LEC cultures with AMPK inhibitor dorsomorphin (compound C) to explore the influence of AMPK on LM-LEC inflammatory profiles. The net effect was a reduction in the expression of COX-2 and HO-1 in LM-LECs. Across the tested isolates, dorsomorphin reduced COX-2 and HO-1 expression by an average of 69% and 83%, respectively (Fig. 4D). Although known to inhibit of NF-κB-dependent inflammatory gene expression, 30 AMPK can stimulate HO-1 and COX-2 expression in endothelial cells.31,32
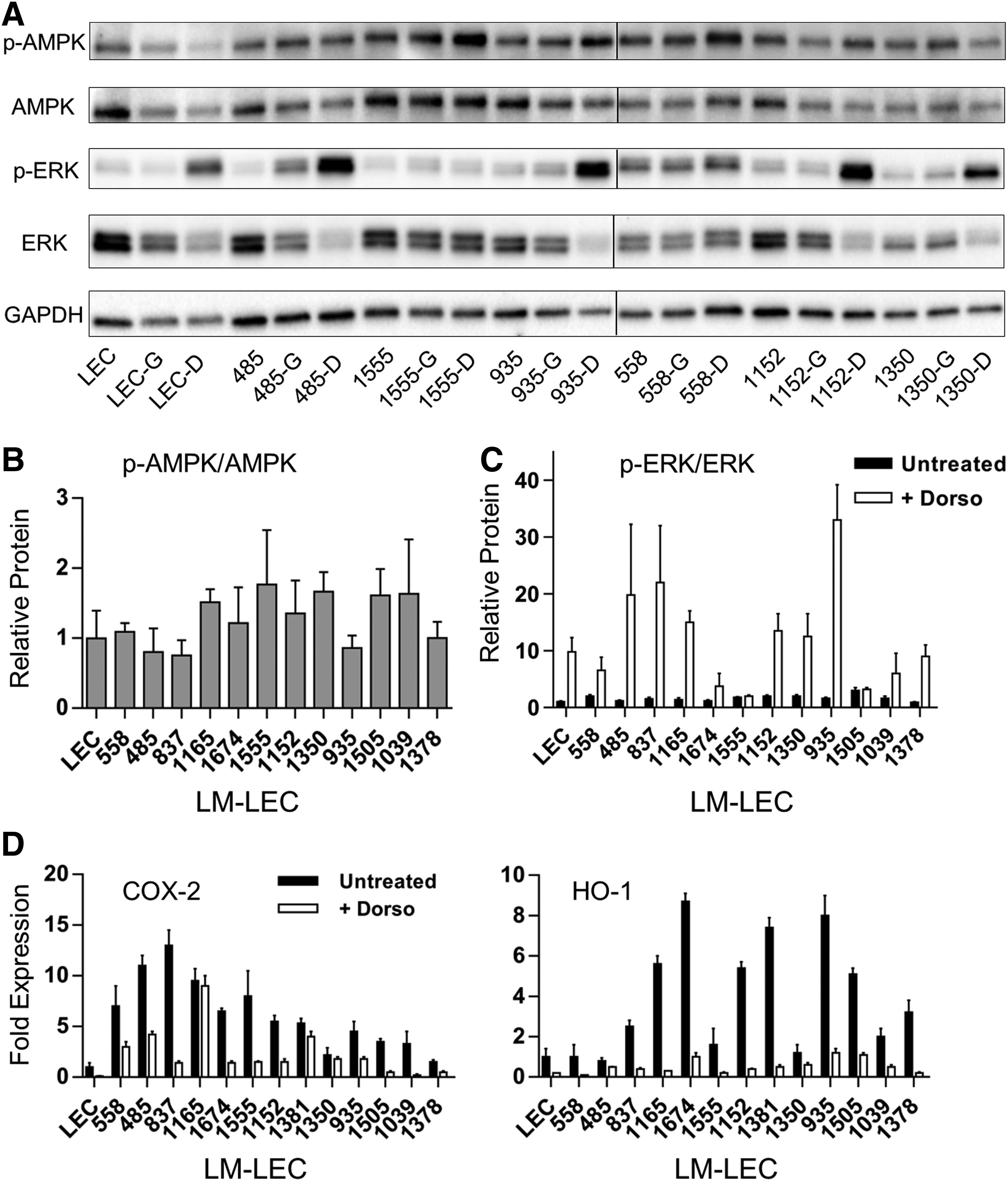
AMPK activity and correlations with ERK1/2 activation and COX-2 and HO-1 gene expression. AMPK activation was determined from ratios of phospho-AMPK to total AMPK protein as measured from immunoblots of triplicate LM-LEC cell samples
Coincidently, we show that dorsomorphin treatment of LM-LECs profoundly enhanced ERK1/2 activity (Fig. 4C). ERK1/2 mediates an alternative, growth/survival signaling pathway that is negatively regulated by AMPK activity.33,34 Apart from demonstrating the efficacy of dorsomorphin, this observation shows that AMPK regulation of inflammation and stress responses may be, in part, ERK1/2 mediated. 35
AMPK drives protective adaptations in response to lipid dysregulation.36,37 Probing a linkage to lipid stress, we measured ANGPTL4 expression in LM-LECs. ANGPTL4 is induced by free fatty acids, 38 regulates lipid metabolism, 39 and protects cells from lipotoxic stress.40,41 ANGPTL4 expression in LM-LECs was measured in parallel with IL-8 to assess the independence of ANGPTL4 gene regulation. ANGPTL4 expression was significantly elevated in all but one LM-LEC population (837 being the exception), averaging 28-fold enhancement over control LECs (11 cultures tested; p < 0.05; Fig. 5A).

Regulation of ANGPTL4 and IL-8 expression in LM-LECs.
IL-8 was less consistently elevated, attaining statistical significance in 6 of the 11 isolates tested. PI3K inhibition had little effect on ANGPTL4 expression (Fig. 5B), differing from IL-8 expression which was generally upregulated in LM-LEC cultures (Fig. 5B). When AMPK was inhibited with dorsomorphin, ANGPTL4 expression was unaltered or reduced. In contrast, IL-8 expression was either unaffected or enhanced upon AMPK inhibition. ANGPTL4 expression, like that of COX-2 and HO-1, was broad in its induction among LM-LEC populations, but the lack of regulation by AMPK, PI3K, and by inference, ERK1/2, suggests a more direct response to cell stress.
Lipotoxic inflammatory gene induction in normal LECs
Defects in lipid metabolism may have a unique impact on LECs given the significance of the lymphatics in systemic lipid management. 42 Scarce research has been devoted to LEC lipid metabolism and lipid tolerance so we exposed dLyNeo cells to different forms of lipid stress and recorded inflammatory responses in terms of the transcriptional induction of COX-2, IL-8, HO-1, VEGFC, ANGPTL4, and sXBP. An unfolded protein response (UPR) effector gene, sXBP, was included as a marker of endoplasmic reticulum (ER) stress. 43 Treatments included exposure of cells to the autophagy inhibitor chloroquine, the fatty acid oxidation (FAO) inhibitor etomoxir, BSA-cPA, and elevated levels of bovine serum (20%). Thapsigargin, an ER stress inducer, was included for comparative purposes. In Figure 6, cell responses are reported relative to untreated cells grown in EGM-2MV. Low serum medium had little impact on targeted gene expression.
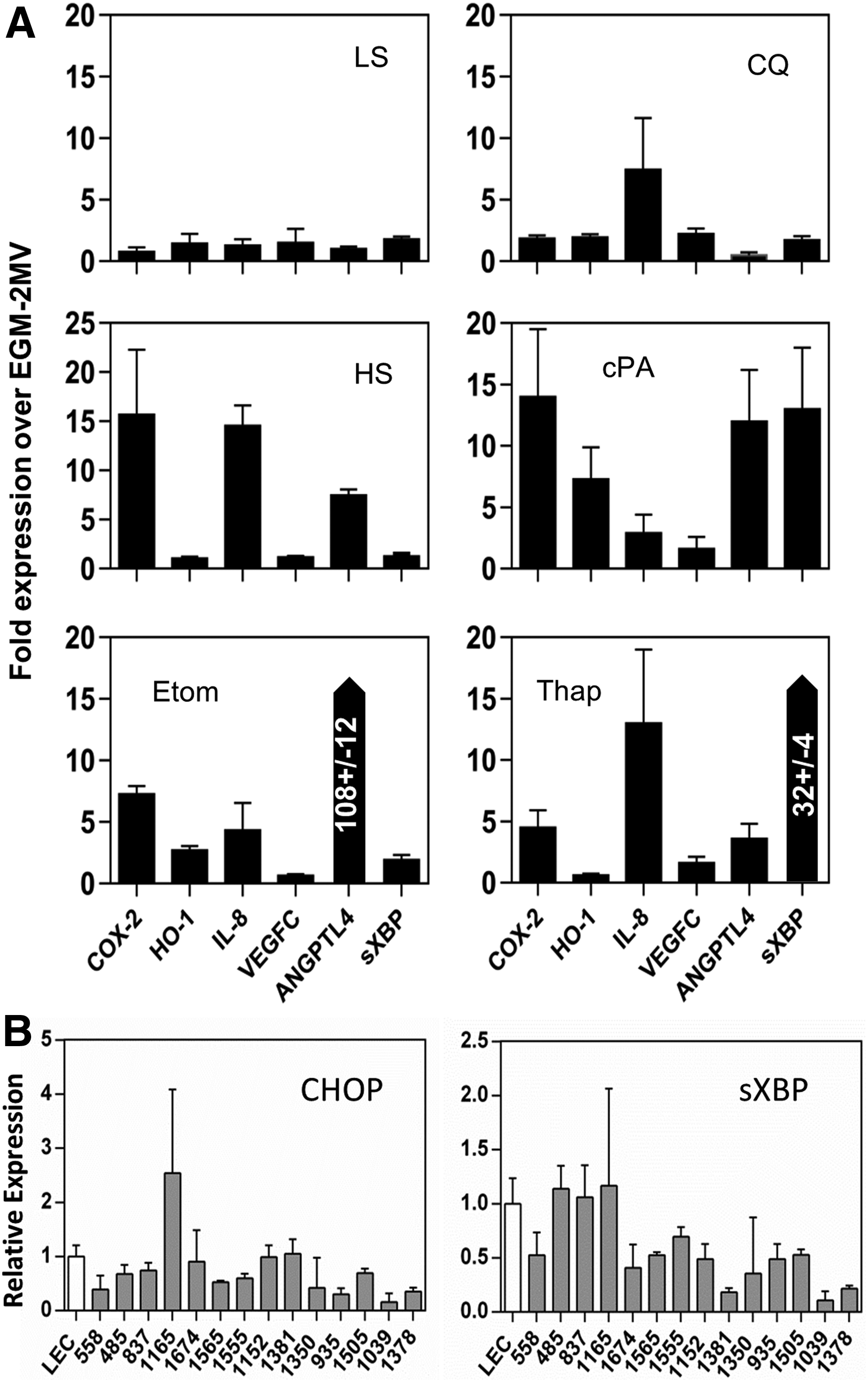
Inflammatory gene expression in lipid stressed LECs. Inflammatory gene expression in control LECs was determined by qRT-PCR following a 24-hour exposure to factors that perturbed lipid homeostasis or cellular metabolism
Thapsigargin, as expected, induced a high level of sXBP (Fig. 6, Thap). IL-8, E-SEL, and COX-2 were also induced by ER stress induction, but HO-1 expression was largely unaffected. Lipotoxic levels of palmitic acid induced all inflammatory genes, including ANGPTL4 and the ER stress marker sXBP. Milder forms of lipid exposure induced patterns of inflammatory gene expression without UPR induction. HS, a source of elevated levels of exogenous lipid, induced COX-2, ANGPTL4, and IL-8 but not HO-1. Inhibition of autophagy (intended to block the metabolism of lipid droplets 44 ), preferentially elevated IL-8 over the other factors. Perturbing endogenous lipid metabolism by inhibiting FAO with etomoxir increased ANGPTL4 expression along with COX-2, HO-1, and IL-8.
Inhibiting fatty acid metabolism was expected to increase intracellular pools of fatty acid, triggering ANGPTL4 induction. Conversely, CQ treatment was expected to reduce fatty acid pools, reducing ANGPTL4 expression, which it accomplished by ∼50%. The response of control LECs to mild lipid stress, and particularly FAO interruption, mimics the reported pattern of inflammatory gene expression in LM-LECs. The similarity can be extended to the absence of VEGFC and sXBP induction in mildly stressed LECs. The former was linked to PI3K activation in LM-LECs (Fig. 2), and the latter was not a feature of LM-LECs. As shown in Figure 6B, transcription of both sXBP and CHOP (UPR genes) was generally indistinguishable from control LECs (Fig. 6B).
Pharmacologic activation of AMPK variably inhibits LM-LEC growth
Investigating the therapeutic potential of AMPK modulation, LM-LECs were exposed to known AMPK modifiers. Agents were evaluated based on specificity and potency, relative to rapamycin, in inhibiting the growth of LM-LEC. Metformin (0.5–2 mM), an indirect activator of AMPK, 45 and dorsomorphin lacked potency or specificity, respectively (data not shown). The AMPK activator AICAR, 46 however, displayed selective efficacy. In Figure 7A, cell growth in the presence of AICAR is reported relative to a baseline of 0% growth (cell stasis) established by treating cells with rapamycin. Four LM-LEC populations displayed a heightened sensitivity to AICAR (isolates 1555, 1152, 935, 1039). Further analysis revealed a correlation between AICAR sensitivity and the ratio of PI3K to AMPK activation (Table 3) in individual cultures. Cultures displaying lower AKT/AMPK activation ratios were those most sensitive to AICAR inhibition (Fig. 7B). This contrasted with rapamycin sensitivity. Rapamycin inhibits mTOR (mammalian target of rapamycin), 47 an effector of PI3K-dependent growth effects. LM-LEC cultures with higher relative PI3K activation were most sensitive to rapamycin (Fig. 7B).
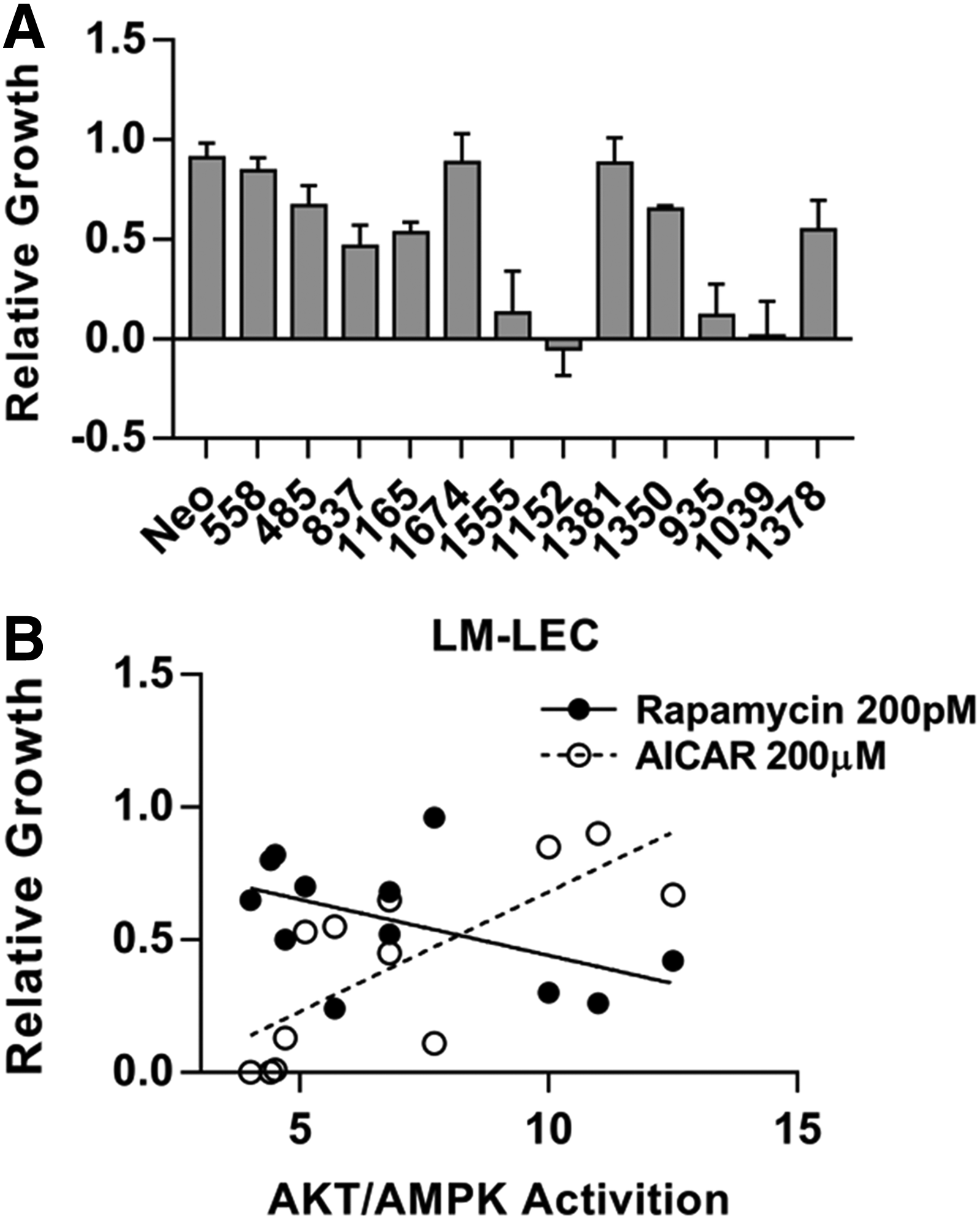
Susceptibility of LM-LECs to AMPK activation. The growth of LM-LEC cultures in response to AMPK activation with AICAR (200 μM) is shown in
Growth in the presence of drug is normalized to growth in its absence. AKT/AMPK activation ratios were determined from phospho-AKT/total AKT and phospho-AMPK/total AMPK ratios calculated from immunoblots. Data were used to generate the linear regression curves shown in Figure 7.
Discussion
Genetic analyses identifying somatic PIK3CA mutations in tissue from LM offer a rational understanding of the dysregulated expansion of lymphatic vessels characteristic of the lesion. Still, variations in the clinical presentation of LM, thought to be a function of developmental timing, anatomical setting, or dispersion of mutated cells, may reflect contributions from other genetic factors. By muting the effects of constitutive PI3K action with a potent inhibitor, we have revealed an inflammatory gene profile in multiple LM-LEC isolates that is independent of activated PIK3CA alleles. Variations in inflammatory profiles and susceptibility to drugs, coupled with the identification of five non-PIK3CA somatic gene variants in LM-LEC populations, implicate unique genetic modifications in the individualization of LM-LECs and possibly their clinical spectrum.
Two candidate mutations present in independent LM-LEC isolates potentially affect lipid storage and cellular lipid homeostasis. FITM2, present in a PROS cell sample, mediates lipid droplet formation at the ER. 22 ATG2A, present in a focal LM, regulates lipid droplet metabolism. 20 A mutation in a third isolate was present in an alternatively spliced form of ANK3. Large ANK3 interacts with the cytoplasmic membrane, but small alternate forms of ANK3, those potentially encoding the variant amino acid substitution, have been localized to endosomes or lysosomes. 27 ANK3-associated lysosomes were shown to function in autophagy-sensitive cellular processes. 28 The remaining two novel alleles identified are gene regulators (MTA1 and TAF1L) previously linked to cancer. However, relevant transcriptional targets have not been fully identified although MTA1 has been associated with VEGF regulation. 48 No variant other than the associated PIK3CA allele was uncovered in the sixth isolate (#837) screened by WES. Each novel allele was detected in LM-LECs at read frequencies comparable to the coexisting PIK3CA allele and was confirmed by direct sequencing of genomic DNA. What effect these alleles have on cell metabolism and growth remains to be proven.
The similarity of LM-LEC inflammatory profiles may be indicative of a common source of cell stress. We have characterized LM-LECs as inflammatory, or activated, based on the upregulation of VEGFC, HO-1, COX-2, and ANGPTL4. Inflammatory cytokines exemplified by IL-8 were less consistently upregulated. VEGFC overexpression appeared independent of inflammatory stress as it was linked to the activation of PI3K. HO-1 overexpression, in contrast, appeared primarily under the regulation of AMPK. COX-2 upregulation by AMPK was apparent although less dramatic. PI3K inhibition enhanced COX-2 expression in some cell populations (Fig. 2) despite the correlation of high COX-2 expression with “strong” PIK3CA alleles (Fig. 3C).
The latter result may indicate an indirect relationship between COX-2 expression and PI3K activity relating to the nature/severity of the underlying metabolic defect. Defects inducing higher COX-2 responses may have required the stronger, more protective, PIK3CA alleles. ANGPTL4 overexpression appeared largely independent of AMPK or PI3K. The induction of ANGPTL4, a regulator of lipid metabolism, strengthens the suspicion that defects in lipid metabolism exist in LM cells.
Lipid droplets are dynamic organelles that store neutral lipids and actively support lipid homeostasis. Ectopic accumulation of LD through nutrient excess or defective turnover contributes to metabolic inflammation and disease.49–52 Ectopic lipid accumulation can also arise from mitochondrial defects and restricted coupling of lipid metabolism to FAO.51,52 Increased fatty acid loads induce ANGPTL4 which, in turn, protects cells by limiting the uptake of triglyceride-derived fatty acid.52,53 Independent of the inhibition of lipoprotein lipase, ANGPTL4 protects against lipotoxic effects such as cell anoikis40,41 and has been linked to cancer growth, spread, and angiogenesis. 54
Systemically, ANGPTL4 partners with AMPK to alter energy usage during physical exertion and stress. 55 Lipid imbalances also induce HO-1. HO-1 is a well-characterized antioxidant, but it may also protect the integrity of cytosolic lipid droplets. 51 In tests to determine the response of control LECs to lipid stress, we observed the overexpression of ANGPTL4 and HO-1 under mild forms of lipid stress. Most notably, the endogenous perturbation FAO in LECs with etomoxir induced the inflammatory gene profile most like LM-LECs. Provocatively, the lone isolate not to overexpress ANGPTL4 (#837) was that for which WES failed to reveal an additional somatic mutation. An expanded investigation of LM-LEC-bearing mutations is required to confirm lipid perturbations as a defining feature of LM.
The preferential selection of activated PIK3CA mutations by LM-LECs may be relevant to the demands of underlying metabolic defects. The demonstration that activated PIK3CA alleles potentially exhibit different levels of intrinsic activity raises the further possibility that specific alleles may be better suited to compensate for mutations that cripple different steps of that metabolic pathway. In the scenario of lipid management, impairments affecting lipid trafficking, storage, or utilization may be differentially accommodated by a PI3K-directed switch from lipid utilization to lipid storage (the promotion of glycolysis). The capacity of PIK3CA alleles to antagonize the growth-limiting activation of AMPK and to compensate for metabolic defects is likely to determine the growth of LM-LECs and the clinical presentation of LM.
The balanced influences of AMPK and PI3K effects on cell stress may be relevant to the efficacy of AMPK activation (with AICAR) in inhibiting LM-LEC proliferation. Whatever the underlying mechanism, modulating AMPK activity may be a complementary therapeutic option to the use of sirolimus.
Coexisting sporadic mutations may influence cell fate determination, playing a role in the clinical presentation of LM and PROS. Our study included three patients with PROS. A “strong” H1047 allele was acquired in two patients with CLOVES. These alleles are not a prerequisite to overgrowth syndromes. 2 In fact, a similar proportion of “strong” (H1047 and H545K) alleles have been linked to CLOVES and localized LM (17/29 and 13/30, respectively; combining our data with Ref. 2 ). The third overgrowth isolate (837) bore the E542K (weak) allele. No additional mutation was discovered in isolate 837, perhaps revealing an exceptional case where a PI3K-related mutation alone underpinned PROS and LM development.
The existence of a unique, undiscovered second mutation has not been ruled out. This patient (837) was diagnosed with Klippel–Trenaunay syndrome (KTS)-like overgrowth. A preponderance of H1047 or H545K alleles is associated with KTS patients (16/20, combined data). The E542 PIK3CA allele present in 837, unopposed by metabolic defect, may be comparable in effect to a “strong” allele. Hence, KTS may be driven by PI3K-activating mutations against a genetic background distinct from that associated with CLOVES and LM and in which PI3K effects are more dominant.
Both CLOVES and KTS overgrowth feature bony and soft tissue hypertrophy, but the characteristic presence of venous malformations in KTS and lipomatous overgrowth in CLOVES distinguishes them. A testable postulate is that a combination of mutations in precursor cells, in addition to timing and anatomical milieu, may specify cell fates unique to PROS and LM.
Testing a multifactorial, and gene-specific, basis to the clinical manifestations of LM and PROS could be undertaken in engineered animal models. Our preliminary results suggest that introducing weak PIK3CA alleles (E542, such as in isolate 837) may be preferable to H1047 alleles. 15 In addition, PIK3CA alleles could be paired with gene variants known to coexist in LM-LECs. Xenograft models may be useful to demonstrate the potential of LM-LECs alone to cause disease and if they do so with disease specificity.
Animal models could be applied to examining the role of LM-LEC elaborated paracrine factors of LM. Specifically, is the recruitment of stem cells a disease component or do paracrine factors stimulate the aberrant growth of neighboring cells? Any factor essential for LM growth may represent a feasible target for treatments (e.g., VEGFC). LECs in which lipid management is crippled may also lead to regional imbalances in lipid loads that impact neighboring cells (e.g., adipose and bone). These influences can be tested in animal models whose design would benefit from an expanded genetic analysis of LM-LECs.
Footnotes
Author Disclosure Statement
No competing financial interests exist.